全面探索肌萎缩侧索硬化症
肌萎缩侧索硬化症(Amyotrophic Lateral Sclerosis, ALS)最初由法国神经学家让-马丁·沙可在1869年描述,但当1939年它迫使著名棒球运动员卢·格里格退役时,在美国的知名度显著增加。在很长一段时间里,ALS被广泛称为卢·格里格病。
1. 什么是肌萎缩侧索硬化症?
"肌萎缩"一词源自希腊语根,意味着肌肉没有营养。它指的是神经细胞向肌肉细胞传递信号的丧失。"侧索"指的是一侧,即脊髓损伤的位置。"硬化"指的是硬化,指的是在晚期ALS中脊髓的硬化特性。
肌萎缩侧索硬化症(ALS)是一种神经退行性疾病,其特征是运动皮层的上运动神经元和脑干及脊髓的下运动神经元逐渐退化,导致随意肌逐渐失去神经支配。到目前为止,还没有治愈ALS的方法。
症状通常首先出现在50至65岁之间。然而,该疾病也可能发生在二三十岁的个体中。最常见的情况是,肌无力通常首先在肢体肌肉中开始,并逐渐进展到近端肌肉。大约三分之一的ALS患者会经历言语障碍、吞咽困难和声音嘶哑等相关症状。在最晚期,ALS患者可能会出现呼吸困难和吞咽困难等症状。
在大多数ALS病例中,患者在首次出现症状后通常存活2-5年,呼吸衰竭是主要的死因。ALS的年发病率为每10万人中有2-3个新病例,患病率约为每10万人中有7-9个病例 [1]。男性比女性更容易患上ALS [2]。
90-95%的ALS病例是散发性的,没有明显的遗传原因,而剩余的5-10%的病例是家族性ALS,表现出相关的遗传显性遗传因素 [3]。散发性(sALS)和家族性ALS(fALS)都与大脑皮层和脊髓运动神经元的退化有关。
2. 肌萎缩侧索硬化症的危险因素
肌萎缩侧索硬化症的病因仍然未知。它可能与遗传和基因缺陷有关 [4]。此外,一些环境因素,如重金属中毒,可能会导致损害。
2.1 遗传因素
分子遗传技术的进步揭示了超过120个潜在的疾病修饰或致病基因与ALS有关。值得注意的是,SOD1、TARDBP、融合肉瘤/脂肉瘤转化蛋白(FUS/TLS)和染色体9开放阅读框72(C9ORF72)显示出最高频率的致病变异,而其他携带此类变异的基因相对不常见 [5]。
| ALS位点编号 | 基因/编码蛋白 | 基因/编码蛋白 | 蛋白功能:疾病机制 |
|---|---|---|---|
| ALS1 | SOD1/Cu-Zn superoxide dismutase | AD (AR) | Dismutates superoxide free radicals: oxidative stress; protein aggregation; mitochondrial dysfunction; axonal transport defects; proteasome impairment; glial dysfunction |
| ALS2 | ALS2/Alsin | AR | Intracellular trafficking |
| ALS4 | SETX/Senataxin | AD | RNA processing |
| ALS5 | SPG11/Spatacsin | AR | Vesicle trafficking; axonal defects |
| ALS6 | FUS/Fused in sarcoma RNA binding protein | AD (AR) | RNA processing; DNA damage repair defects; nucleocytoplasmic transport defects; stress granule function; protein aggregation |
| ALS8 | VAPB/Vesicle-associated membrane protein | AD | Proteasome impairment; intracellular trafficking |
| ALS9 | ANG/Angiogenin | AD | RNA processing |
| ALS10 | TARDBP/TDP-43 | AD | RNA processing; nucleocytoplasmic transport defects; stress granule function; protein aggregation |
| ALS11 | FIG4/Polyphosphoinositide phosphatase | AD | Intracellular trafficking |
| ALS12 | OPTN/Optineurin | AD (AR) | Autophagy; protein aggregation; inflammation; NF-κB regulation; membrane trafficking; exocytosis; vesicle transport; reorganization of actin and microtubules; cell cycle control |
| ALS13 | ATXN2/Ataxin 2 | AD | RNA processing |
| ALS14 | VCP/Valosin-containing protein/ Transitional endoplasmic reticulum ATPase | AD/de novo | Autophagy; proteasome impairment; defects in stress granules; protein aggregation; mitochondrial dysfunction; endoplasmic reticulum dysfunction |
| ALS15 | UBQLN2/Ubiquilin 2 | X-linked AD | Proteasome impairment; autophagy; protein aggregation; oxidative stress; axonal defects |
| ALS16 | SIGMAR1/Sigma non-opioid intracellular receptor 1 | AD and AR | Proteasome impairment; intracellular trafficking |
| ALS17 | CHMP2B/Charged multivesicular body protein 2b | AD | Autophagy; protein aggregation |
| ALS18 | PFN1/Profilin-1 | AD | Axonal defects |
| ALS19 | ERBB4/Receptor tyrosine-protein kinase erbB-4 | AD | Neuronal development |
| ALS20 | hnRNPA1/Heterogeneous nuclear ribonucleoprotein A1 | AD/de novo risk factor | RNA processing |
| ALS21 | MATR3/Matrin-3 | AD | RNA processing |
| ALS22 | TUBA4A/Tubulin α4A chain | AD | Cytoskeleton |
| ALS23 | ANXA11/Annexin A11 | AD | Intracellular trafficking |
| ALS24 | NEK1 | AD | Intracellular trafficking; DNA-damage response; microtubule stability |
| ALS25 | KIF5A/Kinesin heavy chain isoform 5A | AD | Axonal defects; intracellular trafficking |
| ALS-new | GLT8D1/Glycosyltransferase 8 domain-containing protein 1 | AD | Ganglioside synthesis |
| ALS-new | TIA1/Cytotoxic granule-associated RNA-binding protein | AD | Delayed stress granule disassembly; stress granule accumulation |
| ALS-new | C21orf2/Cilia and flagella-associated protein 410 | AD | Microtubule assembly; DNA damage response and repair; mitochondrial function; interacts with NEK1 |
| ALS-new | DNAJC7/DnaJ heat shock protein family (Hsp40) member C7 | Unknown | Protein homeostasis; protein folding and clearance of degraded proteins; protein aggregation |
| ALS-new | LGALSL/Galectin-related protein | Unknown | Unknown |
| ALS-new | KANK1/KN motif and ankyrin repeat domain-containing protein 1 | Unknown | Cytoskeleton; axonopathy |
| ALS-new | CAV1/Caveolin 1 | Unknown | Intracellular and neurotrophic signalling |
| ALS-new | SPTLC1/Serine palmitoyltransferase, long-chain base subunit 1 | AD | Excess sphingolipid biosynthesis |
| ALS-new | ACSL5/Long-chain fatty acid coenzyme A ligase 5 | Unknown | Long-chain fatty acid metabolism |
| ALS-putative | ELP3/Elongator protein 3 | Unknown | Ribostasis; cytoskeletal integrity |
| ALS-putative | DCTN1/Dynactin subunit 1 | AD | Axonal transport |
| ALS-putative | PARK9/Probable cation-transporting ATPase 13A2 | AR | Lysosome function |
| FTD-ALS1 | C9orf72/Chromosome 9 open reading frame 72 | AD | RNA processing; nucleocytoplasmic transport defects; proteasome impairment; autophagy; inflammation; protein aggregation (DPRs) |
| FTD-ALS2 | CHCHD10/Coiled-coil–helix-coiled–coil-helix domain-containing protein 10 | AD | Mitochondrial function; synaptic dysfunction |
| FTD-ALS3 | SQSTM1/Sequestosome-1 | AD | Proteasome impairment; autophagy; protein aggregation; axonal defects; oxidative stress |
| FTD-ALS4 | TBK1/Serine–threonine protein kinase | AD | Autophagy; inflammation; mitochondrial dysfunction |
| FTD-ALS5 | CCNF/Cyclin F | AD | Autophagy; axonal defects; protein aggregation |
表 1. 确定为ALS致病或增加风险的基因
此表信息来源:https://www.nature.com/articles/s41573-022-00612-2
2.2 环境因素
环境因素也在ALS的发病机制中扮演重要角色 [6]。可能的影响因素包括暴露于有毒或传染性病原体、病毒、身体创伤、饮食以及行为和职业因素。
有毒物质:如铅(Pb)和锰(Mn)等重金属中毒。
兴奋性氨基酸和自由基的兴奋导致运动神经元死亡。
研究人员建议,在战争期间或剧烈的体育活动中暴露于铅、杀虫剂和其他环境毒素可能是一些退伍军人和运动员ALS风险增加的原因。
作为一种复杂疾病,遗传和环境因素在ALS的发生和发展中都扮演着重要角色。
2.3 其他危险因素
ALS还与几个潜在的危险因素有关:
- 年龄:尽管ALS也可能发生在年轻人、儿童和老年人身上,但ALS的发病高峰年龄为50至75岁,散发性疾病为58至63岁,家族性疾病为47至52岁,80岁以后发病率迅速下降。
- 性别:男性比女性稍微更有可能发展为ALS。研究表明,总体上,患有该疾病的男性与女性的比例约为1.5。
- 种族和民族:高加索人和非西班牙裔最有可能患有ALS。研究表明,ALS在非洲、亚洲和西班牙裔种族中的发病率可能低于白人。
3. 肌萎缩侧索硬化症的致病机制
尽管经过多年的研究,ALS确切的致病机制仍然未知,疾病的病理生理机制可能源于分子和遗传通路之间复杂的相互作用。科学家们提出了许多致病机制,如下所示:
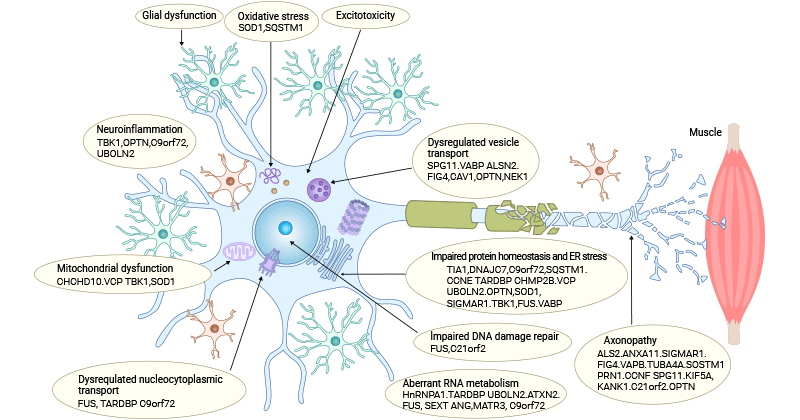
图1. ALS的发病机制
这张图片来源: https://www.nature.com/articles/s41573-022-00612-2
3.1 谷氨酸兴奋毒性
突触释放的谷氨酸增加或从突触间隙中吸收不足导致细胞外谷氨酸水平升高,直接导致谷氨酸受体(包括AMPARs和NMDARs)过度激活 [4]。过度的谷氨酸输入引发兴奋毒性,导致ALS患者皮层过度兴奋和功能障碍。
GLAST和GLT-1,以及它们的星形胶质细胞对应物EAAT1和EAAT2,负责吸收突触谷氨酸以维持最佳细胞外水平,阻止突触间隙中谷氨酸的积累及随后的兴奋毒性 [7]。在胶质细胞中这些谷氨酸转运体的调节失常可能是兴奋毒性及其相关神经病理发生的关键因素。
此外,谷氨酸受体的过度刺激触发细胞质Ca2+的升高,导致细胞内Ca2+流入和积累增加,这是兴奋毒性的一个关键因素。
3.2 氧化应激
氧化应激源于活性氧(ROS)的过度产生与不充分的抗氧化系统之间的不平衡 [8,9]。在ALS中,有许多产生ROS的过程,它们相互联系,形成一个加剧氧化损伤的循环。
线粒体功能障碍可能导致电子传递链中的电子泄漏,从而产生ROS [10,11]。在谷氨酸兴奋毒性期间,ROS也作为副产品产生 [12]。异常的钙水平可以激活诸如NADPH氧化酶之类的酶,它们产生ROS。聚集蛋白(如TDP-43和FUS)的存在有助于氧化损伤。异常的RNA处理可以导致线粒体功能障碍和ROS产生。炎症细胞,如小胶质细胞和星形胶质细胞,作为对疾病的免疫反应的一部分释放ROS。钙调节与线粒体功能障碍和氧化应激密切相关。
与某些家族性ALS病例相关的抗氧化酶突变,特别是超氧化物歧化酶1(SOD1)[13],这些酶的功能障碍降低了细胞中和ROS的能力,导致氧化应激。
ROS通过影响DNA、RNA、脂质和蛋白质等生物分子的功能,以及核酸氧化和脂质过氧化等过程,在神经细胞退化中发挥重要作用。
许多依赖氧气的细胞功能,如信号转导、基因转录、氧化磷酸化和线粒体ATP产生,也通过氧化还原反应产生过氧化氢(H2O2)、超氧阴离子(O2•−)或羟基自由基(HO•)。
自由氧自由基逐渐损害蛋白质、脂质和核酸,导致细胞过程受损、炎症,最终导致细胞死亡。
3.3 线粒体功能障碍
线粒体对神经元的重要性不仅在于其作为通过氧化磷酸化(OXPHOS)产生ATP的众所周知的角色,还在于其参与磷脂生物合成、钙稳态、细胞凋亡和其他生物过程。线粒体在神经元中作为钙缓冲器起着至关重要的作用,调节局部钙动态,例如控制神经递质释放。
在ALS患者中观察到线粒体形态异常、ROS产生增加、线粒体动态缺陷、轴突运输损伤以及线粒体相关膜(MAM)完整性的破坏。线粒体电子传递链(ETC)功能的损害导致ATP产生减少,受损的OXPHOS也会产生高水平的ROS,进一步加速线粒体损伤并最终导致神经元死亡。
与线粒体相关的基因,如SOD1、TDP-43、FUS、C9orf72和CHCHD10,在ALS患者中发生突变。SOD1突变损害了线粒体清除ROS的能力。TDP-43突变异构体干扰线粒体RNA表达,FUS突变异构体与线粒体ATP合酶的催化亚单位相互作用,导致线粒体膜电位异常,减少ATP产生,并影响氧消耗。C9orf72突变提高了神经元Ca2+流入浓度,而polyGR过度表达通过改变线粒体上的蛋白质结合模式诱导线粒体功能障碍。
3.4 蛋白质稳态失衡和异常的RNA代谢
细胞蛋白质稳态(proteostasis)和RNA代谢(ribostasis)在维持大脑结构和功能方面发挥着重要作用。然而,由于衰老、细胞应激和遗传因素,蛋白质稳态和RNA代谢的破坏导致蛋白质错误折叠、不溶性聚集物的沉积以及核糖核蛋白颗粒的异常动态。
大量证据支持蛋白质错误折叠和聚集在ALS发展和进展中的作用。ALS患者表现出三种主要类型的病理聚集物在运动神经元中的独立沉积。大约97%的ALS患者检测到TDP-43、SOD1和FUS阳性包涵体,分别占ALS患者的97%、2%和1% [14]。
SOD1、C9orf72、TDP-43、泛素连接酶2和VCP等基因的突变破坏了UPS和自噬降解途径,导致有毒蛋白质的异常积累,随后导致蛋白质稳态失衡,最终触发细胞死亡。
3.5 核质运输缺陷
核质运输对于细胞核与细胞质之间分子的调节运动至关重要,在ALS中由于C9orf72和FUS等基因的突变而受到破坏 [15]。这些突变影响核孔复合体和核质运输,导致TDP-43等RNA结合蛋白的错位。TDP-43在细胞质中的异常积累是ALS的病理特征。
调节失常的核质运输促成了错误折叠蛋白质的聚集、RNA代谢异常和细胞稳态受损,最终促成了ALS中运动神经元的退化。
3.6 DNA损伤和DNA修复受损
DNA损伤日益被认为是ALS发病机制中的一个因素。在ALS中受影响的主要细胞——运动神经元,由于它们的后有丝分裂性质和高代谢活性,特别容易受到DNA损伤。包括氧化应激、线粒体功能障碍和DNA修复途径受损在内的多种机制可以在ALS中诱导DNA损伤。DNA损伤的积累可以导致基因组不稳定、细胞周期异常和激活细胞死亡途径。此外,涉及DNA修复过程的基因突变,如C9orf72 [16]和FUS [17],也与ALS有关。
3.7 神经炎症
ALS的病理生理与神经炎症过程密切相关,涉及释放可能具有神经保护作用或神经毒性的因素,这些因素促成了运动神经元的病理 [18]。
小胶质细胞和星形胶质细胞在响应ALS相关的损伤和应激时被激活,然后释放促炎分子,包括细胞因子和趋化因子,包括肿瘤坏死因子α(TNF-α)、白细胞介素1β(IL-1β)和白细胞介素6(IL-6),这些分子可以放大免疫反应并促成神经元损伤。外周免疫细胞如T淋巴细胞的浸润和NF-κB途径的激活也会引起神经炎症。
神经炎症与ALS中的氧化应激相互关联。被激活的胶质细胞释放的炎症介质可以诱导氧化应激,反之,氧化应激也可以激活炎症途径,形成一个自我持续的炎症循环。失调的血脑屏障和突变蛋白相互作用进一步加剧神经炎症,导致运动神经元损伤和ALS疾病进展。
3.8 少突胶质细胞功能障碍
少突胶质细胞是负责髓鞘化轴突的胶质细胞,确保高效的神经信号传递。在ALS患者的脊髓前角灰质中观察到病理性包涵体、髓鞘异常、脱髓鞘,甚至少突胶质细胞退化 [19,20]。
此外,与ALS相关的基因如SOD1、FUS和TDP-43的突变对少突胶质细胞产生不利影响,触发蛋白质聚集体的形成,诱导内质网应激,并最终导致细胞凋亡 [20]。有重要证据表明,活化的小胶质细胞和星形胶质细胞对少突胶质细胞及其前体细胞具有细胞毒性作用。谷氨酸介导的兴奋毒性和氧化应激也是少突胶质细胞退化的原因。
3.9 轴突运输中断
轴突运输是维持神经元存活的基本过程。鉴于控制各种运动蛋白的基因突变,如KIF5A、DCTN1和DYNC1H1,与ALS有关 [21,22],轴突运输受损被认为是一种病理机制。SOD1、FUS和TDP-43的突变也改变了轴突运输。
除了在编码轴突运输相关蛋白的基因中发生的突变外,ALS发病机制中的轴突运输缺陷可能还涉及几个过程,包括破坏微管稳定性 [23]、运动蛋白的过度磷酸化[24]以及货物与运动蛋白之间联系的减弱。
4. 肌萎缩侧索硬化症的生物标志物
ALS的标记物在预测疾病发展、评估预后和确定治疗策略中起着至关重要的作用。ALS标记物主要包括RNA结合蛋白(RBPs)、与ALS相关的基因和非编码RNA。
| 生物标志物 | 描述 | |
|---|---|---|
| RBPs | TDP-43 | ALS患者细胞质蛋白包涵体的主要成分;异常磷酸化、泛素化、溶解和/或核耗竭是ALS的突出病理特征 |
| FUS | FUS蛋白聚集体在ALS患者中很常见 | |
| TAF15 | TAF15在散发性和家族性ALS患者中发生突变;TAF15积累会加速神经退行性变 | |
| EWSR1 | EWSR1蛋白在散发性ALS患者中呈现弥漫分布或点状颗粒结构;野生型EWSR1的过度表达会导致神经退行性变 | |
| ATXN2 | ATXN2中间长度多谷氨酸的扩展增加了ALS的风险;ALS中ATXN2三核苷酸重复扩增可以预测疾病风险 | |
| HnRNPs | HnRNPs在ALS中相对罕见,但肯定参与了ALS的发病机制,可能通过与其他常见病理基因(TDP-43)的结合 | |
| MATR3 | 在ALS患者中发现MATR3部分错位;MATR3基因的S85C错义突变是ALS的遗传原因 | |
| TIA1/TIAR | TIA1/TIAR是重要的应激颗粒组分;TIAR可能参与缺血后神经元细胞死亡,而在ALS患者中发现了TIA1 LCD突变的增加风险 | |
| ALS-related genes | SOD1 | SOD1是一种强效的抗氧化酶;至少发现170种SOD1基因突变会导致ALS;突变SOD1的毒性可能由最初的错误折叠引起,与ALS的发病机制有关 |
| C9orf72 | C9orf72中的异常GGGGCC六核苷酸重复扩增被认为是最常见的家族性ALS遗传原因 | |
| CHCHD10 | CHCHD10是一种位于线粒体膜间隙的线粒体蛋白; CHCHD10突变似乎是ALS的相对罕见原因,并且在被诊断为额颞叶痴呆的患者中可能更常见 |
|
| TBK1 | TBK1是核因子-κB激酶家族的成员;TBK1的突变是ALS/FTD共病(10.8%)的主要遗传原因,而与单独的ALS(0.5%)关联较少 | |
| TUBA4A | TUBA4A的突变与家族性ALS相关,所有TUBA4A突变患者都经历了伴随上运动神经元和下运动神经元迹象的脊髓发作 | |
| NEK1 | 确定了NEK1功能丧失变异与散发性ALS风险之间的显著关联 | |
| C21orf2 | C21orf2与ALS相关;超过75%的突变被发现可能是有害的 | |
| CCNF | CCNF的突变可能增加TDP-43聚集体并导致ALS的发作;CCNF变异被认为是ALS的罕见原因,不同地区人群的变异率不同 | |
| KIF5A | KIF5A是与ALS相关的新基因;KIF5A C末端货物结合尾域的突变导致ALS | |
| ANXA11 | ANXA11的突变可以通过涉及异常蛋白聚集的增益功能机制参与ALS的发病机制 | |
| GLT8D1 | 糖基转移酶的活性被认为与ALS的发展有关,特别是家族性ALS | |
| SPG11 | SPG11的突变被认为是导致常染色体隐性遗传和青少年ALS特征的痉挛性截瘫的致病因素 | |
| Non-coding RNA | miRNAs | miR-27a、miR-34a、miR-124、miR-142-5p、miR-155和miR-338-3p被研究作为与ALS相关的生物标志物和潜在治疗靶点 |
| lncRNAs | NEAT1_2可以在ALS早期调节与ALS相关的RNA结合蛋白的功能;在带有FUS、TDP-43和SOD1突变的ALS患者中,共发现了20种反义lncRNA | |
| circRNAs | 通过抑制DBR1的功能调节circRNAs生物生成被认为是ALS的潜在治疗策略;hsa_circ_0023919、hsa_circ_0088036和hsa_circ_0063411是ALS的潜在基于血液的生物标志物 | |
| Others | UA | 血清UA水平与ALS患者死亡风险之间存在负相关 |
| CL | CL水平的变化也可能反映在几个ALS模型中观察到的线粒体完整性的丧失 | |
| CHIT1 | 已指出ALS患者脑脊液中CHIT1水平升高 | |
| NfL | 血清NfL与疾病进展呈正相关,而较高的NfL水平表明生存期较短 | |
表2. ALS的生物标志物
此表信息引用自: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8300638/
5. 肌萎缩侧索硬化症的最新研究
一方面,科学家正在研究与ALS相关的遗传变异。早期研究专注于寻找单个基因的变异与这种疾病之间的联系。随着技术的不断进步,科学家现在可以更准确地观察整个基因组,从而取得了一些有趣的发现,例如在C9ORF72基因中发现的变异。
另一方面,研究人员正在研究神经元死亡的机制。神经元死亡是ALS的标志之一,但对此过程知之甚少。然而,一些新的研究正在揭示神经元死亡的机制,这可能有助于开发更好的治疗方法。
目前,国际上正在尝试使用神经营养因子、抗氧化剂如维生素E和维生素C,以及肌酸(Creatine)、辅酶Q10(CoQ10)等与利鲁唑(Rilutek)联合使用,为ALS提供保护性治疗。然而,上述治疗尚未在临床试验中得到证实。
一些研究也在探索不同的治疗方法,例如基因治疗和免疫疗法。基因治疗的理念是将人工修饰的基因引入体内,以修复或替换受损的基因。免疫疗法旨在利用身体的免疫系统来识别并攻击导致肌肉萎缩的细胞。虽然这些方法仍处于研究的早期阶段,但它们可能为未来的治疗提供线索。
总之,ALS是一种致命的瘫痪性神经退行性疾病,影响上运动神经元和下运动神经元,导致逐渐加重的肌无力、痉挛和过度反射。死亡通常发生在症状发作后的2-5年内,主要是由于呼吸肌减弱导致的呼吸衰竭。到目前为止,还没有治愈或有效治疗ALS的方法,尽管某些药物被用来缓解症状。然而,已经鉴定出多个与ALS相关的基因,研究这些基因及其涉及的过程可能为ALS治疗提供策略。
参考文献:
[1] Hardiman, O.; Al-Chalabi, A.; et al. Amyotrophic Lateral Sclerosis [J]. Nat. Rev. Dis. Primers 2017, 3, 17085.
[2] Manjaly, Z.R.; Scott, K.M.; et al. The Sex Ratio in Amyotrophic Lateral Sclerosis: A Population Based Study [J]. Amyotroph. Lateral Scler. 2010, 11, 439–442.
[3] Byrne S, Walsh C, Lynch C, et al. Rate of familial amyotrophic lateral sclerosis: a systematic review and meta-analysis [J]. J Neurol Neurosurg Psychiatry, 2011, 82(6):623-627.
[4] Duan QQ, Jiang Z, et al. Risk factors of amyotrophic lateral sclerosis: a global meta-summary. Front Neurosci [J]. 2023 Apr 24;17:1177431.
[5] Boylan K (2015). Familial amyotrophic lateral sclerosis [J]. Neurol Clin 33:807–830.
[6] Ahmed A, Wicklund M P. Amyotrophic Lateral Sclerosis: What Role Does Environment Play? [J]. Neurologic Clinics, 2011, 29(3):689-711.
[7] Pajarillo, E., Rizor, A., et al. (2019). The role of astrocytic glutamate transporters GLT-1 and GLAST in neurological disorders: potential targets for neurotherapeutics [J]. Neuropharmacology 161:107559.
[8] Cunha-Oliveira T., Montezinho L., et al. Oxidative Stress in Amyotrophic Lateral Sclerosis: Pathophysiology and Opportunities for Pharmacological Intervention [J]. Oxid. Med. Cell Longev. 2020;15:5021694.
[9] Motataianu A, Serban G, Barcutean L, Balasa R. Oxidative Stress in Amyotrophic Lateral Sclerosis: Synergy of Genetic and Environmental Factors [J]. Int J Mol Sci. 2022 Aug 19;23(16):9339.
[10] Kausar S., Wang F., Cui H. The Role of Mitochondria in Reactive Oxygen Species Generation and Its Implications for Neurodegenerative Diseases [J]. Cells. 2018;7:274.
[11] Greco V., Longone P., et al. Crosstalk Between Oxidative Stress and Mitochondrial Damage: Focus on Amyotrophic Lateral Sclerosis [J]. Adv. Exp. Med. Biol. 2019;1158:71–82.
[12] Olloquequi, J., Cornejo-Córdova, E., et al. (2018). Excitotoxicity in the pathogenesis of neurological and psychiatric disorders: therapeutic implications [J]. J. Psychopharmacol. 32, 265–275.
[13] Higgins C.M., Jung C., et al. Mutant Cu, Zn superoxide dismutase that causes motoneuron degeneration is present in mitochondria in the CNS [J]. J. Neurosci. 2002;22:RC215.
[14] Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis [J]. Neuron 2013;79:416–438.
[15] Vanneste J, Van Den Bosch L. The Role of Nucleocytoplasmic Transport Defects in Amyotrophic Lateral Sclerosis [J]. Int J Mol Sci. 2021 Nov 10;22(22):12175.
[16] Balendra R, Isaacs AM. C9orf72-mediated ALS and FTD: multiple pathways to disease [J]. Nat Rev Neurol. 2018;14:544–558.
[17] Vance C, Rogelj B, Hortobágyi T, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6 [J]. Science. 2009;323:1208–1211.
[18] Fritz E, Izaurieta P, et al. Mutant SOD1-expressing astrocytes release toxic factors that trigger motoneuron death by inducing hyperexcitability [J]. J Neurophysiol. 2013;109(11):2803–14.
[19] Kang SH, Li Y, Fukaya M, et al. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis [J]. Nat. Neurosci. 16(5), 571–579 (2013).
[20] Annelies Nonneman , Wim Robberecht, Ludo Van Den Bosch. The role of oligodendroglial dysfunction in amyotrophic lateral sclerosis [J]. Neurodegener Dis Manag. 2014;4(3):223-39.
[21] Nicolas A, et al. Genome-wide analyses identify KIF5A as a novel ALS gene [J]. Neuron. 2018;97(6):1268–1283.
[22] Munch C, et al. Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS [J]. Neurology. 2004;63(4):724–726.
[23] Godena, V. K., Brookes-Hocking, et al. (2014). Increasing microtubule acetylation rescues axonal transport and locomotor deficits caused by LRRK2 roc-COR domain mutations [J]. Nat. Commun. 5:5245.
[24] Morfini, G. A., Bosco, D. A., et al. (2013). Inhibition of fast axonal transport by pathogenic SOD1 involves activation of p38 MAP kinase [J]. PLoS One 8:e65235.





