“颤抖的”帕金森病
帕金森病(PD)最初由英国医生詹姆斯·帕金森在1817年的《关于震颤麻痹的论文》中描述,他描述了六位患有一种以消瘦和震颤为特征的神经系统障碍的患者 [1]。七十年后,法国神经学家让-马丁·沙可首次对这种疾病进行了临床描述,并以詹姆斯·帕金森的名字命名,以表彰其贡献。
1. 什么是帕金森病?
帕金森病(PD)是一种慢性、进展性的神经系统疾病,已成为全球第二常见的神经退行性疾病,影响65岁以上人口的1% [2]。
帕金森病主要分为散发性和家族性两种形式。大多数(90-95%)患者患有散发性PD,也称为特发性PD(IPD),这是由个体遗传构成与环境因素之间复杂的相互作用决定的。家族性病例,由遗传突变的孟德尔遗传导致,约占帕金森病患者的5-10%。携带与PD相关基因的个体有更高的PD风险。
2. 帕金森病的症状有哪些?
帕金森病的症状通常逐渐开始,最初较轻。与该疾病相关的有多种症状,但其进展和严重程度在个体间有所不同。帕金森病患者很少遇到所有或大多数这些症状。
2.1 帕金森病的运动症状
帕金森病的核心运动症状包括肌强直和震颤、肌僵直、运动迟缓、姿势不稳和行走或步态困难。
- 震颤,手、臂、腿和颌部的颤抖。
- 肌强直,四肢或躯干的紧张或僵硬。
- 运动迟缓,动作缓慢,面部表情减少或面具脸,眨眼频率减少,平衡和协调能力受损。
- 姿势不稳,平衡问题。
- 行走或步态困难,如犹豫、过渡和运动开始时的冻结步态。
- 言语问题,声音低沉或声音变软,可能开始时很强然后逐渐消失,可能失去声音的常态量和情感变化;在晚期,言语可能会加快,导致单词拥挤,可能出现口吃。
2.2 帕金森病的非运动症状
- 认知障碍
- 早饱
- 自主功能障碍,如便秘和直立性低血压
- 疲劳 [3]
- 幻觉 [4]和妄想
- 神经精神障碍,如抑郁、焦虑、冷漠、易怒和睡眠障碍
- 感觉症状,嗅觉功能障碍
- 泌尿系统问题(紧急、频繁、失禁)
- 性问题,如勃起功能障碍
- 视力问题
- 体重减轻
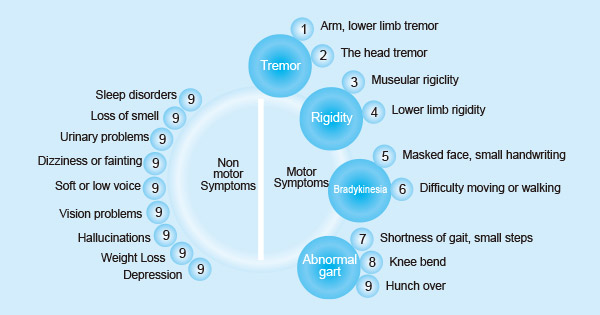
图1. 帕金森病的症状
除了运动症状外,PD患者通常还必须应对相关的非运动症状。这强调了PD不仅仅是中枢神经系统的疾病,而是一种系统性疾病。
3. 帕金森病的原因是什么?
帕金森病的原因尚不完全清楚,但大量研究表明,帕金森病的病理是黑质(SN)多巴胺能神经元的退化和含有α-突触核蛋白(α-syn)聚集体的Lewy体的存在 [5-7]。α-突触核蛋白的聚集和积累使神经元退化持续下去。
研究还表明,帕金森病的病因与多种因素有关,包括遗传易感性、环境因素、衰老等。
3.1 遗传因素
至少5%的PD病例与多个基因的突变有关,包括富含亮氨酸的重复激酶2(LRRK2)、α-突触核蛋白(SNCA)、β-葡萄糖苷酶1(GBA1)、磷酸酶和张力蛋白同源物诱导激酶1(PINK1)、帕金(PRKN)和PARK7(DJ-1)[8-10]。这些基因与家族性PD的高风险有关。
这些突变基因的产物导致细胞内运输异常、氧化应激、线粒体功能障碍、蛋白质质量控制中断、蛋白质错误折叠和聚集、泛素-蛋白酶系统功能障碍以及激酶活性改变,在PD的发病机制中起着关键作用。某些突变还可能导致PD的早期发病。
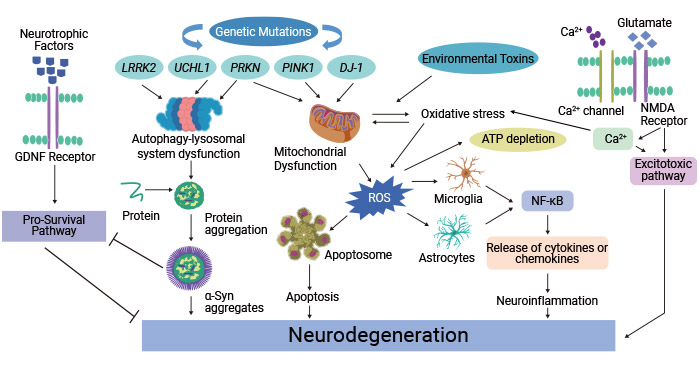
图2. 帕金森病病理生理中涉及的分子机制
这张图片引用自:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8962417/
| PARK基因座 | 基因/蛋白 | 遗传方式 | 生理功能 |
|---|---|---|---|
| PARK1/4 | SNCA/α‐synuclein | Autosomal dominant (AD) | 囊泡介导的内吞作用 神经递质释放 外泌体释放 分子伴侣介导的自噬 α-突触核蛋白聚集和累积是Lewy体的主要成分,导致神经元退化 |
| PARK5 (putative) | UCHL1 | AD | 泛素C末端水解酶L1:处理泛素化蛋白和泛素前体 |
| PARK8 | DARDARIN/LRRK2 | AD | 囊泡介导的内吞作用 自噬 神经递质释放 内溶酶体运输 外泌体释放 单基因致病突变可能降低GTP酶活性或增加激酶活性 |
| PARK11(putative) | GIGYF2 | AD | GRB10相互作用的GYF蛋白2:抑制翻译启动 |
| PARK13(putative) | HTRA2 | HtrA丝氨酸肽酶2:蛋白水解活性,促进细胞凋亡 | |
| PARK17 | VPS35 | AD | 自噬 内溶酶体运输 高尔基体复合体运输 VPS35突变导致逆行体功能障碍、蛋白聚集、线粒体功能障碍、多巴胺信号传导受阻、质膜受体循环和囊泡运输受阻[11] |
| PARK18 | EIF4G1 | AD | 控制编码线粒体、细胞存活和生长蛋白的mRNAs的翻译启动 |
| PARK21 | DNAJC13/RME‐8 | AD | 囊泡介导的内吞作用 内体排序/运输 致病性DNAJC13突变导致异常的内体α-突触核蛋白滞留 |
| PARK22 | CHCHD2 | AD | 涉及线粒体呼吸链复合体;CHCHD2的T61I突变导致自主显性PD形式 |
| PARK2 | PRKN/Parkin | Autosomal recessive (AR) | 囊泡介导的内吞作用 线粒体质量控制、神经递质释放 泛素化、调节线粒体自噬降解 |
| PARK6 | PINK1 | AR | 线粒体质量控制(线粒体自噬、融合和分裂、线粒体衍生囊泡) |
| PARK7 | PARK7/DJ‐1 | AR | 线粒体功能 线粒体活性氧的转录调控 |
| PARK9 | ATP13A2 | AR | 内溶酶体通路 外泌体释放 与早发性PD相关的Kufor-Rakeb综合征(KRS);ATP13A2突变导致溶酶体功能障碍,引起α-突触核蛋白异常堆积[12] |
| PARK14 | PLA2G6 | AR | 膜运输 磷脂代谢 线粒体功能 |
| PARK15 | FBX07 | AR | 泛素化和蛋白酶体降解、增殖 |
| PARK19 | DNAJC6/Auxilin | AR | 囊泡介导的内吞作用 高尔基体-溶酶体运输 |
| PARK20 | SYNJ1/Synaptojanin 1 | AR | 囊泡介导的内吞作用 突触自噬 |
| PARK23 | VPS13C | AR | 涉及线粒体生物发生和线粒体自噬的脂质转运蛋白 |
表信息来源:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6138432/ 和 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10308076/
3.2 环境因素
- 神经系统问题:核上性麻痹、威尔逊病、亨廷顿病、哈勒·瓦尔登-施帕茨综合征和阿尔茨海默病也可能导致帕金森病。
- 脑损伤:导致意识水平变化的创伤性脑损伤,增加了受伤后几年患PD的风险 [13]。
- 居住地区:PD的地理分布存在差异,这可能是由于环境和遗传因素的差异所致。
- 职业:某些职业类别或职位与PD的高发病率有关。工作压力增加患帕金森病的风险。
- 农药暴露:农药(包括赛克宁和六氯环己烷)在与帕金森病相关的所有化学物质暴露中报告最为一致。
- 接触金属:职业性接触各种金属被认为与PD的发展有关。
3.3 衰老
年龄是患帕金森病的最大风险因素,60岁以后发病率显著增加,在随后的几十年中呈指数增长。帕金森病主要影响老年人,尤其是55至65岁之间的人群,65岁以上人群中有1%,85岁以上人群中有5%或更多的人受影响 [14]。
早发性帕金森病(YOPD)发生在50岁以下的人群中。在极少数情况下,儿童和青少年可能出现类似帕金森病的症状。被诊断为YOPD的人有更频繁的帕金森病家族史和更长的生存期。
4. 关于帕金森病的数据和事实
4.1 数据
流行病学显示,患病率为15~328/10万,大约1%的65岁以上人群。每年的发病率是10到21/10万人口。在美国,发病率是每10万人中21例 [15]。鉴于全球预期寿命的增加,预计到2030年,受帕金森病影响的人数以及随之而来的个人、社会和经济负担将急剧增加 [16]。到2020年,美国将有近100万人患有帕金森病,每年约有6万名美国人被诊断出帕金森病。全球有超过1000万人患有帕金森病。
4.2 事实
年龄:帕金森病主要发生在老年人中,尽管年轻人也可能患上帕金森病,但估计只有4%的帕金森病患者在50岁之前被诊断。帕金森病的平均发病年龄是60岁,PD的患病率随年龄增长而增加。
性别:男性比女性患帕金森病的可能性高1.5倍。有帕金森病父母或兄弟姐妹的人可能因遗传突变而使患帕金森病的风险加倍。
种族:患病率存在种族差异,白人最高,其次是黄种人,黑人最低。

图3. 帕金森病的事实
4.3 帕金森病的案例
当名人公开他们的疾病时,这有助于提高人们对疾病的认知,增加他们对疾病的了解。
穆罕默德·阿里(1984年诊断)在离开拳击运动三年后被诊断出患有帕金森病。他在全球范围内提高了人们对帕金森病的认识,并帮助在亚利桑那州凤凰城建立了穆罕默德·阿里帕金森中心。
迈克尔·J·福克斯(1991年诊断)是世界上最著名的帕金森病患者之一。他致力于帕金森病研究的进一步发展,并建立了迈克尔·J·福克斯帕金森病研究基金会。
布赖恩·格兰特(2008年被诊断出患有帕金森病)并成立了布赖恩·格兰特基金会,以帮助其他人做同样的事情。
此外,乔治·H·布什、比利·康诺利、艾伦·阿达、尼尔·戴蒙德和帕特·托尔皮也被诊断出患有帕金森病。
5. 分子通路在帕金森病病理生理中的作用
尽管帕金森病的确切原因尚未完全理解,但有几个分子通路和过程被认为在这种疾病的发展和进展中起着作用。了解这些通路对于开发旨在减缓或停止帕金森病进展的靶向治疗至关重要。
5.1 多巴胺系统功能障碍
病理上,PD主要表现为黑质致密部(SNPC)投射到纹状体的多巴胺能神经元的减少,沿黑质纹状体通路。黑质纹状体通路是一个重要的多巴胺能通路,负责调节自愿运动和运动功能。
这种神经退行性导致纹状体中多巴胺水平显著降低,这增加了基底神经节中的整体兴奋性驱动,阻断了自愿运动控制并引起帕金森病特有的运动障碍[17]。当多巴胺能神经元损失高达50%至80%时,患者通常开始表现出帕金森病的运动症状。
5.2 α-突触核蛋白聚集
α-突触核蛋白是一种由140个氨基酸组成的突触前蛋白,具有多种构象,并以多种寡聚态存在,维持着动态平衡。突变的α-突触核蛋白会经历构象变化,使其易于形成聚集体和Lewy体。α-突触核蛋白的聚集被认为具有神经毒性,并促成了黑质中多巴胺能神经元的进行性退化。这种神经退行性导致了帕金森病的特征性运动症状。
5.3 线粒体功能障碍
与PD相关的线粒体功能障碍基因包括SNCA、LRRK2、VPS35、CHCHD2、PARK、PINK1、DJ-1、ATP13A2、PLA2G6和FBXO7的突变。PINK1和Parkin突变与线粒体自噬有关,可能导致缺陷线粒体的积累并促成功能障碍。
PD中的线粒体功能障碍导致能量产生缺陷(特别是在多巴胺能神经元中)、增加的氧化应激、线粒体DNA损伤、细胞凋亡、改变的钙稳态、受损的线粒体自噬、α-突触核蛋白积累,以及整体细胞功能障碍。所有这些最终加速了多巴胺能神经元的退化和死亡。
5.4 氧化应激
氧化应激是PD发病机制中的关键因素。在PD中,这种氧化损伤对黑质中的多巴胺能神经元尤为有害。线粒体功能障碍、α-突触核蛋白聚集和炎症促成了PD中活性氧(ROS)的增加产生。氧化应激可以进一步加剧线粒体损伤,触发细胞凋亡途径,并促进α-突触核蛋白的错误折叠。
5.5 炎症和免疫反应
神经炎症是恢复正常大脑结构和功能的保护机制。然而,当过度激活时,神经炎症成为神经退行性变的重要驱动因素。慢性神经炎症是PD病理学中一个关键的标志。
神经炎症的特征是大脑中的免疫细胞如小胶质细胞和星形胶质细胞的激活。免疫反应是由α-突触核蛋白聚集、线粒体功能障碍和氧化应激等因素触发的。激活的小胶质细胞释放炎症介质,促成受影响区域(尤其是黑质)的慢性炎症环境。这种持续的免疫反应可以加剧神经元损伤,并促进PD的进展。
5.6 自噬功能障碍
自噬在清除错误折叠的蛋白(特别是α-突触核蛋白,其聚集是PD病理的标志)中起着至关重要的作用。神经元,特别是PD中受影响的多巴胺能神经元,严重依赖有效的自噬来维持细胞稳态。自噬功能障碍可能损害神经元健康,并促成PD中观察到的进行性退化。
与自噬相关的基因突变,如PINK1和Parkin,已与家族性PD形式相关联。这些基因对于维持正常的自噬功能至关重要。自噬依赖于溶酶体来降解吞噬的细胞物质。溶酶体途径的功能障碍可以损害自噬过程,并促成PD中有毒蛋白聚集的积累。
总结来说,PD是一种进展性的神经退行性疾病,病理特征是黑质致密部多巴胺能神经元的丧失和含有α-突触核蛋白的Lewy体的形成。多种因素共同促成了PD的发展,包括形成Lewy体的α-突触核蛋白聚集、线粒体功能障碍、神经炎症,以及兴奋毒性和金属积累。
目前对帕金森病的研究强调了遗传和环境因素之间的相互作用、PD病理从外周向中枢神经系统的传播,以及对神经炎症和多巴胺神经元退化分子机制的深入探索。
参考文献:
[1] Parkinson T. Outlines of Zoonosological Tables. Lond. Med. Phys. J. 1817;38:449–453.
[2] Tysnes, O.-B.; Storstein, A. Epidemiology of Parkinson's Disease [J]. J. Neural Transm. 2017, 124, 901–905.
[3] Siciliano Mattia, Trojano Luigi, Santangelo Gabriella. et al. Fatigue in Parkinson's disease: A systematic review and meta-analysis [J]. Mov. Disord, 2018, undefined: undefined.
[4] Swann Peter, O'Brien John T. Management of visual hallucinations in dementia and Parkinson's disease [J]. Int Psychogeriatr, 2018, undefined: 1-22.
[5] Trist B., Hare D., Double K. (2019). Oxidative stress in the aging substantia nigra and the etiology of Parkinson's disease [J]. Aging cell 18:e13031.
[6] Alvarez-Erviti L., Seow Y., et al. (2011). Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission [J]. Neurobiol. Dis. 42 360–367. 10.1016/j.nbd.2011.01.029.
[7] Roberts R., Wade-Martins R., Alegre-Abarrategui J. (2015). Direct visualization of alpha-synuclein oligomers reveals previously undetected pathology in Parkinson's disease brain [J]. Brain 138 1642–1657.
[8] Kim J., Daadi E.W., Oh T., Daadi E.S., Daadi M.M. Human Induced Pluripotent Stem Cell Phenotyping and Preclinical Modeling of Familial Parkinson's Disease [J]. Genes. 2022;13:1937.
[9] Verstraeten A., Theuns J., Van Broeckhoven C. Progress in unraveling the genetic etiology of Parkinson disease in a genomic era [J]. Trends Genet. 2015;31:140–149.
[10] Kalinderi K., Bostantjopoulou S., Fidani L. The genetic background of Parkinson's disease: Current progress and future prospects [J]. Acta Neurol. Scand. 2016;134:314–326.
[11] Rahman AA, Morrison BE. Contributions of VPS35 Mutations to Parkinson's Disease [J]. Neuroscience. 2019 Mar 1;401:1-10.
[12] Zhang F, Wu Z, Long F, et al. The Roles of ATP13A2 Gene Mutations Leading to Abnormal Aggregation of α-Synuclein in Parkinson's Disease. Front Cell Neurosci. 2022 Jul 6;16:927682.
[13] Gardner Raquel C, Byers Amy L, Barnes Deborah E, et al. Mild TBI and risk of Parkinson disease: A Chronic Effects of Neurotrauma Consortium Study [J] .Neurology, 2018, 90: e1771-e1779.
[14] Pringsheim T, Jette N, Frolkis A, et al. The prevalence of Parkinson's disease: a systematic review and meta-analysis [J]. Movement Disorders Official Journal of the Movement Disorder Society, 2015, 29(13):1583-1590.
[15] Savica R, Grossardt BR, Bower JH, et al. Incidence and pathology of synucleinopathies and tauopathies related to parkinsonism [J]. JAMA Neurol, 2013, 70:859-66.
[16] Dorsey ER, Constantinescu R, Thompson JP, et al. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030 [J]. Neurology, 2007; 68:384-6.
[17] Gordián-Vélez WJ, Chouhan D, et al. Restoring lost nigrostriatal fibers in Parkinson's disease based on clinically-inspired design criteria [J]. Brain Res Bull. 2021 Oct;175:168-185.





