细胞因子和类风湿性关节炎
类风湿关节炎(RA)是一种典型的自身免疫性疾病,全球发病率为1%。如果患者没有得到及时诊断和治疗,2年治残率达到70%,平均寿命将缩短10-15年。因此,RA一直是全世界的研究热点。
近年来,随着分子生物学和免疫学的迅速发展,RA的临床治疗取得了很大的突破。免疫细胞和各种细胞因子成为了RA发病机制的研究重点。细胞因子是参与RA免疫炎症反应的重要分子,不仅为RA的发生机制提供了更深入的理论认识,也为临床免疫治疗奠定了坚实的基础。本文总结了RA治疗中具有前景的细胞因子,包括以下几个部分。
什么是类风湿性关节炎?
类风湿关节炎(RA)是一种慢性自身免疫性炎性疾病,以免疫炎症反应引起的关节受损进而导致关节功能障碍和残疾为特征,严重影响患者的生活质量。RA的关节症状通常包括关节疼痛、关节肿胀、关节僵硬、关节功能丧失和畸形。症状从轻度到重度不等。了解RA的早期症状将帮助医疗服务团队为患者提供更好地治疗。RA往往会影响到其他组织的正常功能,包括肺部、心脏和眼睛。
RA的诊断通常包括症状分析,身体检查,X射线表现和实验室检查。早期诊断RA的最佳时间是在症状出现后6个月内,疾病诊断和有效的治疗,将大大的减缓或阻止疾病的发展,减少RA的破坏性影响。
类风湿性关节炎的发病机制是什么?
RA是一种自身免疫性的慢性炎症性关节疾病,以免疫球蛋白G(IgG)和瓜氨酸蛋白(ACPA)的自身抗体为特征[1]。如果治疗不及时,RA可导致关节损伤和残疾[2]。主要是因为抗瓜氨酸蛋白抗体(ACPA)与含瓜氨酸的抗原可形成免疫复合物,随后与类风湿因子(RF)结合,导致丰富的补体激活。
ACPAs能与许多自身蛋白上的瓜氨酸残基结合,包括Vimentin、α-烯醇酶、纤维蛋白、纤维蛋白原、组蛋白和II型胶原蛋白。目前,这些免疫反应激活的组织尚不确定。(图1)[3] [4]。
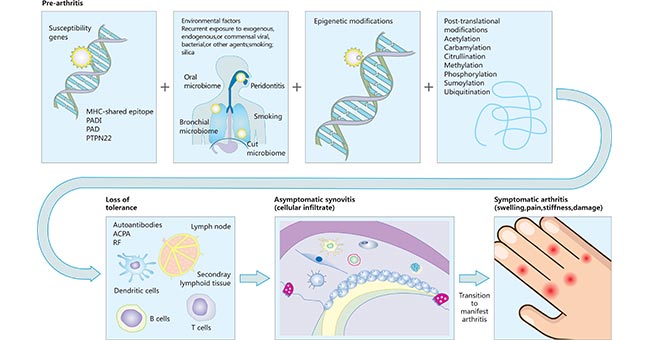
图1. 类风湿性关节炎的发病途径
*图片来源于Lancet 的出版物
参与类风湿关节炎的细胞因子
众所周知,细胞因子是由免疫细胞分泌的细胞分子,参与免疫反应中细胞与细胞之间的信息传递,并刺激细胞向炎症、感染和创伤的部位移动。在RA中,细胞因子可分为四类。促炎细胞因子(IL-13和IL-20)和天然细胞因子拮抗剂(IL-1ra和sTNF-RI)[5] [6] [7] [8]。如前所述,本文只关注在RA治疗中具有前景的细胞因子,包括IL-1、IL-6、IL-17、IL-23、VEGF和TNF-α。
IL-1
关节组织中IL-1主要由滑膜软骨细胞产生。IL-1是破坏RA关节软骨最重要的促炎细胞因子之一。IL-1能刺激人滑膜细胞中磷脂酶A2(PLA2)和环氧酶-2(COX-2)的表达,提高其活性,然后分解卵磷脂膜产生花生四烯酸,导致PGE2的产生和释放[9]。IL-1能促进滑膜细胞和软骨细胞中前列腺素(PGE2)和胶原酶的合成和释放。IL-1可诱导RA关节的滑膜细胞增生,刺激滑膜细胞产生蛋白激酶。
PGE2和胶原醇引发滑膜炎症和软骨基质崩解。局部免疫复合物和游离胶原等分解产物刺激IL-1分泌,加重RA。蛋白激酶直接作用于软骨基质,引起软骨吸收。IL-1还可导致变性蛋白酶的合成和分泌,引起间质降解和骨质破坏。IL-1诱导滑膜和软骨合成大量的金属蛋白酶-胶原和基质蛋白。基质蛋白影响软骨基质降解,或促进蛋白多糖产生[10]。
IL-6
IL-6是一类具有广泛作用的炎症细胞因子,它对适应性和先天性免疫细胞都有影响。此外,它在破骨细胞分化中起作用,可导致骨质破坏,介导慢性炎症引起的贫血。IL-6的表达失调是RA发病的一个重要因素。许多研究表明,RA患者血清中高表达IL-6[11]。进一步发现,IL-6的水平往往在滑膜液内最高,滑膜细胞是RA中IL-6的主要来源[12]。
在过去的十年中,IL-6抑制剂是治疗RA较为有效的生物制剂 [13]。托珠单抗(Tocilizumab)是全球首个针对IL-6的人源化单克隆抗体。Tocilizumab能够破坏IL-6与可溶性IL-6受体的结合。
IL-17
IL-17俗称IL-17A,是IL 17家族的创始成员,由A-F六个配体组成。最初,研究者发现IL 17的受体是IL17R,进一步研究表明,IL17的受体IL17R由复合物IL-17RA和IL 17RC两个亚单位组成[14, 15]。Th17细胞是IL17的主要来源。IL17影响各种细胞类型的活性,导致类风湿性关节炎,引发软骨、骨炎症和骨破坏。
IL17在小鼠膝关节中过量表达可诱导关节炎症、骨侵蚀和软骨蛋白多糖丢失。IL17和IL-1有相同的分解代谢特征,但IL17代谢较慢。有趣的是,通过与TNF的协同作用,IL17的代谢速度明显提高。IL17可直接抑制软骨细胞合成基质,产生降解软骨基质的酶,具有双重分解作用 [16, 17]。
IL-23
IL-23是一种异源性细胞因子,由它与Il-12共用的IL-12p40和IL-23p19亚基组成。IL-23可被IL-23受体识别,受体由IL-23A和Il-12β1组成。大量证据表明,IL-23在Th17细胞的分化和RA的发病过程中起着重要作用[18]。动物模型研究表明,清除IL23,可诱导Th17细胞数量减少,预防关节炎。IL-23可抑制Th17分化,预防疾病发生,但一旦疾病发生,就很难阻止 [19]。
VEGF
1994年,Fava等通过免疫组化发现RA患者滑膜组织中表达VEGF,染色阳性的细胞主要是滑膜内层细胞。从RA患者滑膜组织的原代细胞培养中获得大量成纤维细胞(FLS)和少量巨噬细胞。这些FLS细胞能分泌大量的VEGF。临床研究证实,RA患者外周血中VEGF值越高,牵连的关节数量越多,关节病变越严重。有研究者认为,外周血中VEGF水平可作为反映RA患者病情程度的评价指标,也可作为血沉的评价指标。
此外,许多细胞因子也参与VEGF的调节。IL-1和TNF-α均可通过上调VEGF的分泌,促进滑膜血管生成。相关实验证实,联合使用IL-1和TNF-α抗体培养RA的滑膜细胞,可使VECF的表达降低45%,但仅使用其中一种抗体对阻断滑膜细胞VECGF的表达无明显效果。
TNF-α
在RA中,TNF-α主要由患者外周血单核细胞和关节滑膜巨噬细胞分泌,TNF-α通常在关节病理组织中表达,50%的RA患者在关节滑膜中检测到TNF。
TNF-α主要参与RA发病的三个病理过程。第一,TNF-α可增加血管内皮细胞粘附分子的表达,使血液中的白细胞通过与粘附分子的相互作用而集中到关节腔内。第二,TNF-α可刺激结缔组织细胞和多形核细胞产生前列腺素等小分子,并作为炎症的介质。第三,TNF-α刺激滑膜细胞和软骨细胞,减少破骨细胞的糖蛋白合成,增加糖蛋白降解,并产生胶原酶和其他中性蛋白酶释放骨钙,导致骨和软骨的破坏。在RA患者的免疫治疗中,使用TNF-α-mAb或可溶性TNF受体可明显改善患者的临床症状。
参考文献:
[1] Josef S Smolen, Daniel Aletaha, et al. Rheumatoid arthritis [J]. Lancet. 2016.
[2] Josef S Smolen, Daniel Aletaha, et al. Rheumatoid arthritis [J]. Nat Rev Dis Primers. 2018.
[3] Anquetil F, Clavel C, et al. IgM and IgA rheumatoid factors purified from rheumatoid arthritis sera boost the Fc receptor-and complement-dependent effector functions of the disease-specific anti-citrullinated protein autoantibodies [J]. J Immunol. 2015, 194: 3664–74.
[4] Girbal-Neuhauser E, Durieux JJ, et al. The epitopes targeted by the rheumatoid arthritis-associated antifi laggrin autoantibodies are posttranslationally generated on various sites of (pro)filaggrin by deimination of arginine residues [J]. J Immunol. 1999, 162:585–94.
[5] Kasama, T., Isozaki, T., et al. Clinical effects of tocilizumab on cytokines and immunological factors in patients with rheumatoid arthritis [J]. Int.Immunopharmacol. 2016, 35: 301–306.
[6] Jimenez-Boj, E. et al. Interaction between synovial inflammatory tissue and bone marrow in rheumatoid arthritis[J].J. Immunol. 2005, 175:2579–2588.
[7] Choy, E. Understanding the dynamics: pathways involved in the pathogenesis of rheumatoid arthritis [J]. Rheumatology 2012, 51:3-11.
[8] Ponchel, F., Goéb, V., et al. An immunological biomarker to predict MTX response in early RA [J].Ann.Rheum.Dis. 2014, 73(11): 2047–2053.
[9] Ghezzi P, Cerami A. Tumor necrosis factor as a pharmacological target [J]. Methods Mol Med. 2004, 98 :1-8.
[10] Niki Y, Yamada H, et al. Membrane-associated IL- 1contributes to chronic svnovitis and cartilare destruction in human IL-1 alpha transgenic mice [J]. J Immunol. 2004, 72 (1) 577-584.
[11] Baumann, H. & Kushner, I. Production of interleukin-6 by synovial fibroblasts in rheumatoid arthritis [J]. Am.J. Pathol. 1998, 152, 641–644.
[12] Stone, J. H. et al. Trial of Tocilizumab in Giant-Cell Arteritis [J].N. Engl. J. Med.2017, 377, 317–328.
[13] Viet L. Bui, Ernie Brahn.Cytokine targeting in rheumatoid arthritis [J]. Immunol.2019, 29: 613-621.
[14] Gaffen, S. L. An overview of IL17 function and signaling [j]. Cytokine.2008, 43, 402–407.
[15] Maitra, A. et al. Distinct functional motifs within the IL17 receptor regulate signal transduction and target gene expression [J]. Proc.Natl Acad.Sci. USA. 2007, 104, 7507–7511.
[16] Geboes, L.et al. Proinflammatory role of the TH17 cytokine IL22 in collageninduced arthritis in C57Bl/6 mice [J].Arthritis Rheum.2009, 60, 390–395.
[17] Wim B. van den Berg and Pierre Miossec. Il 17 as a future therapeutic target for rheumatoid arthritis [J]. Nat Rev Rheumatol. 2009, 5: 549-553.
[18] Lubberts, E. The IL-23-IL-17 axis in inflammatory arthritis [J]. Nature Reviews Rheumatology. 2015, 11, 415–429.
[19] Cornelissen, F. et al. IL-23 Dependent and Independent Stages of Experimental Arthritis:No Clinical Effect of Therapeutic IL-23p19 Inhibition in Collagen-induced Arthritis [J]. PLoS One. 2013, 8.





