内质网主导的细胞凋亡
日期:2018-01-11 14:03:23
在上一章中我们综合阐述了细胞凋亡的概念,以及详细探讨了细胞凋亡的内部线粒体途径,本篇文章将带来内质网主导的细胞凋亡机理,希望有助于科学探讨。
1.细胞凋亡的概念
2.细胞凋亡的分类
2.1细胞凋亡的内部线粒体途径
2.2细胞凋亡的内部内质网途径
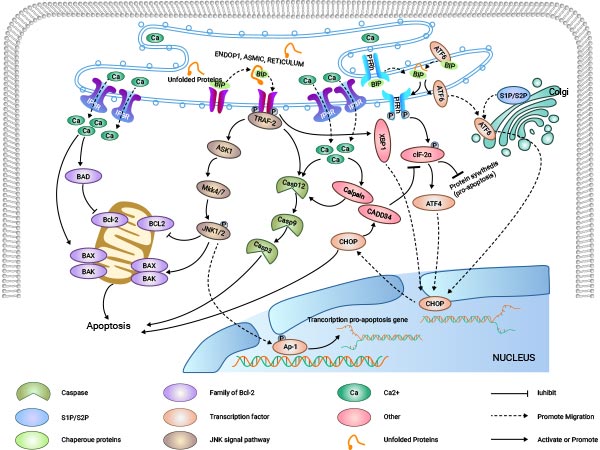
内质网是蛋白质合成的主要加工厂,也是Ca2+重要的储存库。因此,内质网在维持细胞Ca2+离子的稳定、蛋白的合成、加工中起到关键性作用。内质网腔内Ca2+离子失衡、错误折叠或未折叠蛋白增多,则会引起内质网的应激反应(endoplasmic reticulum stress,ERS)。内质网应激反应可减少细胞中蛋白质的合成、增加蛋白正确折叠、维持Ca2+稳态,但过度的应激反应会触动细胞内的凋亡信号,促使细胞凋亡。
2.2未正确折叠蛋白介导的细胞凋亡
在真核生物体内,为正确折叠蛋白反应(unfolded protein response,UPR)是细胞对抗内质网应激的一种重要的自我保护机制。当细胞中出现长时间或高强度的UPR时,三种内质网上的跨膜蛋白PERK、IREI、ATF6在发挥修复作用的同时,也可以同时启动由ERS介导的三种细胞凋亡途径。
2.2.1.1PERK信号通路
PERK是分布于内质网膜上的一种蛋白激酶。当蛋白正常折叠时,其与分子伴侣如BIP/GRP78相结合形成稳定的复合物;当有蛋白未正常折叠时,未正确折叠的蛋白与BIP/GRP78相结合,竞争性干扰 Bip/Grp78与PERK的相互作用。释放的PERK通过低聚化和反向自身磷酸化的方式被活化,活化的PERK可以使翻译起始因子2的α亚单位(eIF-2α)磷酸化。在应激反应的早期磷酸化的eIF2α抑制蛋白质的翻译与合成,减轻内质网中蛋白折叠的负荷量,从而对细胞起到保护作用;随着应激反应时间和强度的增加,磷酸化的eIF-2α诱导激活转录因子ATF4的转录表达,ATF4可以促进凋亡信号分子CHOP/ GADD153的表达,进而促进细胞凋亡。
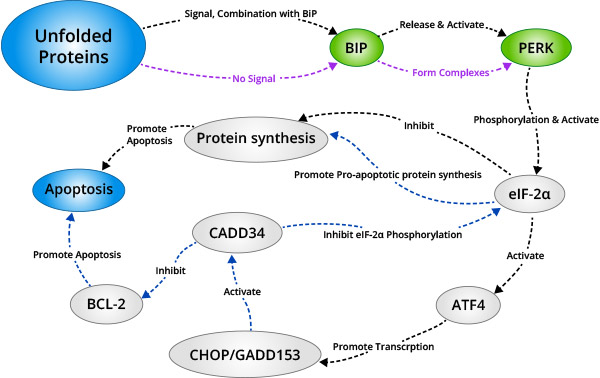
2.2.1.2IREI信号通路
IREI是分布于内质网膜上的另一种蛋白激酶。该信号通路的激活方式与PERK的激活方式相同。当未正常折叠蛋白在内质网中累积时,IREI-BIP/GRP78复合物解离,释放的IREI寡聚化和反向自身磷酸化后被激活;激活的IREI可以传导细胞存活信号和细胞凋亡信号。在凋亡的过程中,激活的IRE1招募胞浆调节蛋白TRAF-2,间接招募和激活c—Jun N端激酶,激活的c—Jun N端激酶通过磷酸化抑制Bcl-2家族中抑制凋亡蛋白的活性,促进蛋白凋亡;另一方面激活的TRAF-2同时激活Caspase12,启动Caspase级联反应,介导细胞凋亡;此外IRE1还具有核糖核酸酶活性,切割XBP1mRNA,促进XBP1mRNA成熟,增强分子伴侣蛋白和CHO的转录表达,从而促进细胞凋亡。
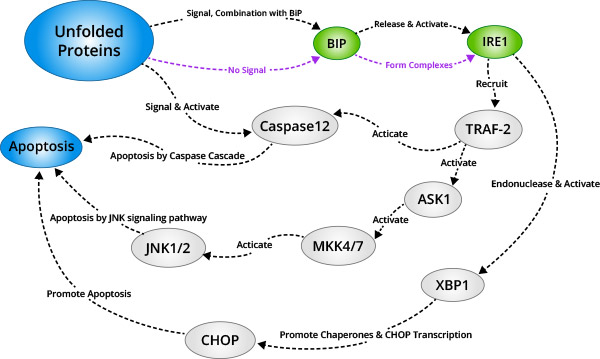
2.2.1.3ATF6信号通路
ATF6是内质网膜上的II型跨膜蛋白。ATF6的N端胞内区域包含了b-ZIP的DNA转录激活域和核定位信号。在非应激状态下,以酶原的形式分布于内质网膜上。当在内质网应激状态下,ATF6以囊泡的方式向高尔基体转移。在高尔基体中被S1P 和S2P剪切激活,然后再核定位信号的牵引下迁移至细胞核,在细胞核中诱导包括CHOP/ GADD153在内的内质网应激基因的转录表达。
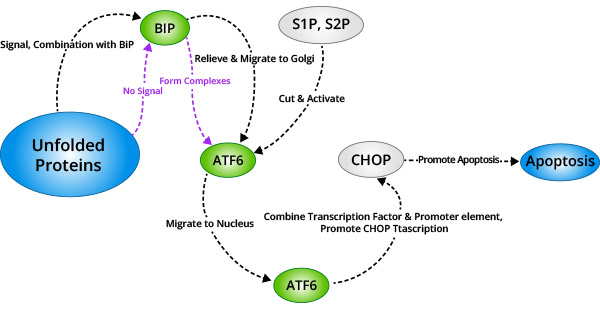
PERK、IRE1、ATF6三条信号通路均可对诱导产生CHOP/GADD153,CHOP/GADD153的激活是内质网应激反应的直接结果,CHOP/GADD153在生长停止和细胞死亡中起到重要作用。
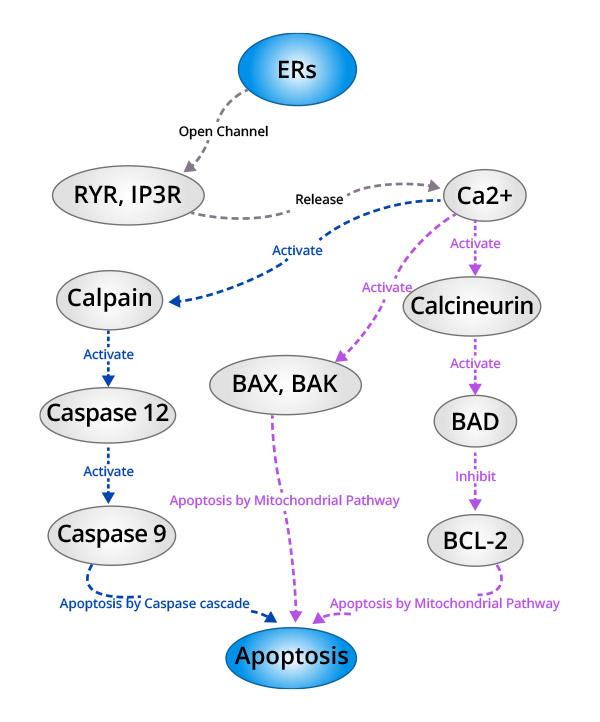
2.2.2Ca2+失衡介导的细胞凋亡
在细胞正常运转情况下,内质网主要通过RyR和IP3R通道将内质网腔内的Ca2+释放入胞质,通过钙泵将胞内的Ca2+泵入内质网腔中,从而维持内质网Ca2+稳态。当内质网接收到应激信号时,内质网内的Ca2+稳态被打破,大量的Ca2+进入胞内和线粒体内,一方面影响线粒体以及Bcl-2家族蛋白的活性,使细胞走向凋亡,另一方面激活胞内的中性半胱氨酸内肽酶Calpain,活化的Calpain可能通过激活Caspase级联反应影响细胞凋亡。
往期回顾:线粒体主导的细胞凋亡
下期预告:死亡受体主导的细胞凋亡
3.参考文献
[1] Green D R, Kroemer G. The pathophysiology of mitochondrial cell death [J]. Science, 2004, 305: 626-629. Groenendyk J, Michalak M. Endoplasmic reticulum quality control and apoptosis [J]. Acta Biochimica Polonica, 2005, 52(2): 381-395.
[2] Bastida-Ruiz D, Aguilar E, Ditisheim A, et al. Endoplasmic reticulum stress responses in placentation - A true balancing act [J]. Placenta, 2017, 57: 163-169.
[3] Kang-sheng LIU, Zheng-hang PENG, Weng-jun CHENG, et al. Endoplasmic reticulum stress-induced apoptosis in the development of reproduction [J]. Reproductive and Developmental Medicine, 2016, 27(1): 51-59.
[4] Li J, Lee B, Lee A S. Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and NOXA by p53 [J]. Journal of Biological Chemistry, 2006, 281(11): 7260-7270.
[5] Burton G J, Yung H W, Murray A J. Mitochondrial - Endoplasmic reticulum interactions in the trophoblast: Stress and senescence [J]. Placenta, 2017, 52: 146-155.
[6] Marchi S, Patergnani S, Missiroli S, et al. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death [J]. Cell Calcium, 2017.
[7] Breckenridge D G, Germain M, Mathai J P, et al. Regulation of apoptosis by endoplasmic reticulum pathways [J]. Oncogene, 2003, 22(53): 8608-8618.










