PAH Recombinant Monoclonal Antibody
-
货号:CSB-RA169000A0HU
-
规格:¥1320
-
图片:
-
其他:

产品详情
-
Uniprot No.:P00439
-
基因名:
-
别名:Phenylalanine-4-hydroxylase (PAH) (EC 1.14.16.1) (Phe-4-monooxygenase), PAH
-
反应种属:Human
-
免疫原:A synthesized peptide derived from human PAH
-
免疫原种属:Homo sapiens (Human)
-
标记方式:Non-conjugated
-
克隆类型:Monoclonal
-
抗体亚型:Rabbit IgG
-
纯化方式:Affinity-chromatography
-
克隆号:17D10
-
浓度:It differs from different batches. Please contact us to confirm it.
-
保存缓冲液:Rabbit IgG in phosphate buffered saline, pH 7.4, 150mM NaCl, 0.02% sodium azide and 50% glycerol.
-
产品提供形式:Liquid
-
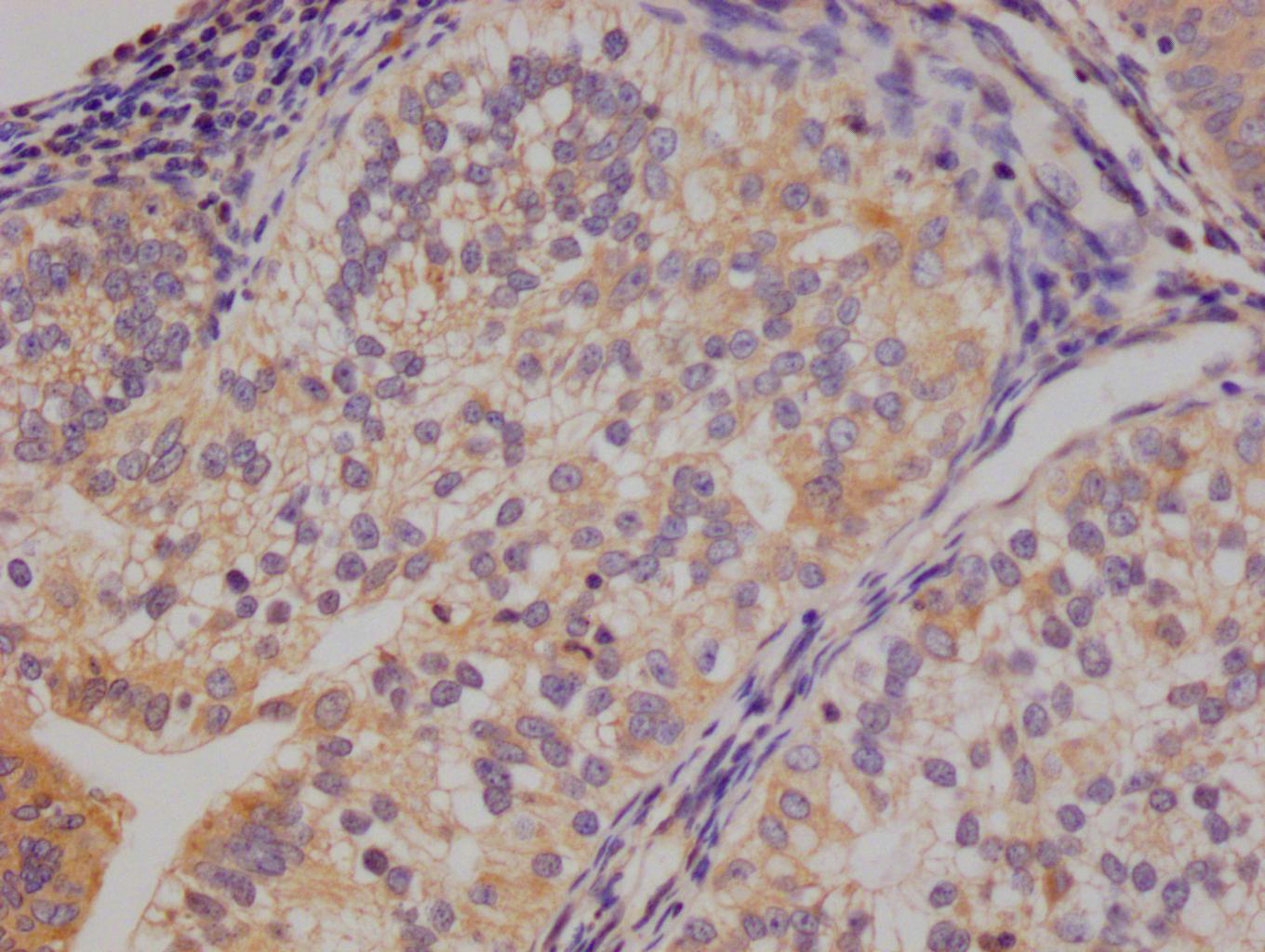
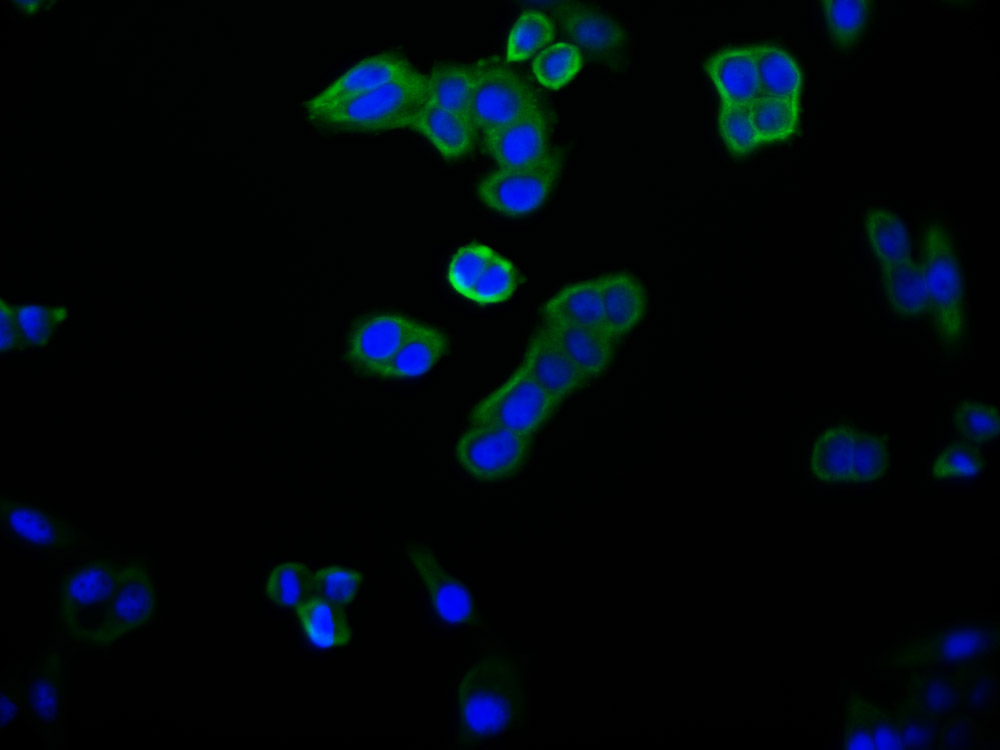
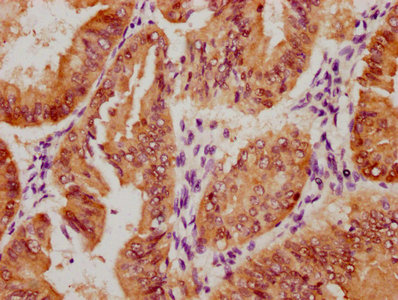
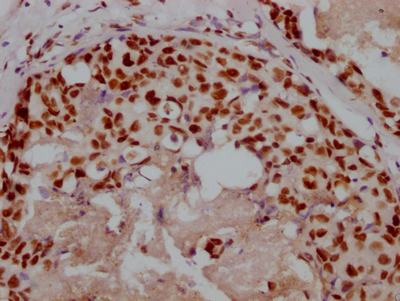
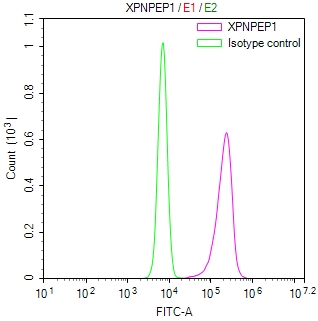
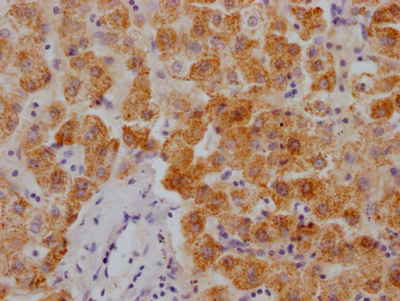
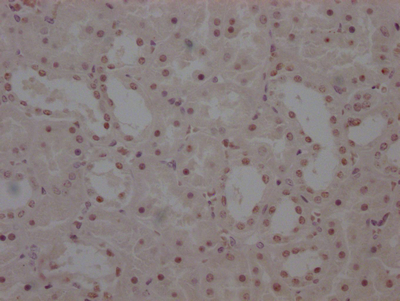
应用范围:ELISA, IHC, IF
-
推荐稀释比:
Application Recommended Dilution IHC 1:50-1:200 IF 1:50-1:200 -
Protocols:
-
储存条件:Upon receipt, store at -20°C or -80°C. Avoid repeated freeze.
-
货期:Basically, we can dispatch the products out in 1-3 working days after receiving your orders. Delivery time maybe differs from different purchasing way or location, please kindly consult your local distributors for specific delivery time.
产品评价

相关产品
靶点详情
-
功能:Catalyzes the hydroxylation of L-phenylalanine to L-tyrosine.
-
基因功能参考文献:
- Molecular diagnostics for DNAJC12 variants are thus mandatory in all patients in which deficiencies of PAH and BH4 are genetically excluded. PMID: 29174366
- Mutational spectrum of the phenylalanine hydroxylase gene in patients with phenylketonuria in the central region of China has been reported. PMID: 29390883
- a spectrum of PAH mutations complied from 35 PKU children who are all Chinese Han population from north Jiangsu, is reported in this study. PMID: 29413232
- strong association between particular mutations and minihaplotypes could be useful for prenatal diagnosis and preimplantation genetic diagnosis in affected families PMID: 28676969
- PAH mutation was associated with hyperphenylalaninemia. PMID: 29032371
- Report a novel intron 11 regulatory element, which is involved in exon 11 splicing, as revealed by the investigated pathogenic effect of variants c.1199+17G>A and c.1199+20G>C, identified in PKU patients. Both mutations cause exon 11 skipping in a minigene system. PMID: 29684050
- three novel variants of the PAH gene, p.E178K (c.532G>A), p.V245M (c.733G>A), p.S250F (c.749C>T), showed impaired protein expression and enzyme activity. PMID: 29653233
- Among phenylketonuria patients, some with autism at the time of evaluation, six mutations were identified: p.E280K, p.G352Vfs, IVS10nt11, p.I224T, p.R261Q, and p.R252W. Study found no correlation between autism and mutations affecting the phenylalanine hydroxylase gene, but the age of die PMID: 26759449
- Studies involving co-expression of differently phenylalanine hydroxylase (PAH) alleles have shown that one variant form can influence the other when assembled into a tetramer and have effect on enzyme activity in vitro, and on BH4 responsiveness and PKU phenotype in patients carrying them. [review] PMID: 26919687
- 17 VUS (37%; 7 in ACADM, 9 in GALT, and 1 in PAH) were reclassified from uncertain (6 to benign or likely benign and 11 to pathogenic or likely pathogenic). We identified common types of missing information that would have helped make a definitive classification and categorized this information by ease and cost to obtain PMID: 27308838
- Our findings contribute to better understanding of structure and function of PAH mutated enzymes and optimal treatment of PKU patients carrying these mutations using BH4 supplementation. PMID: 28653649
- We obtained a PAH gene variant spectrum for the Northern Chinese population and devised a strategy for gene diagnosis using phenylketonuria pedigrees. PMID: 28982351
- Phenylalanine hydroxylase gene mutations are associated with phenylketonuria. PMID: 28604955
- DNA methylated alleles of the phenylalanine hydroxylase promoter remodeled at elevated phenylalanine levels in newborns with hyperphenylalaninemia have been characterized. PMID: 28389235
- In this study, we assigned the phenotypic outcome of three of the five novel mutations and furthermore six not previously classified mutations to one of the four PKU categories. PMID: 26542770
- PAH mutation analyses provided further support for genotype-phenotype correlations in patients with hyperphenylalaninemia. The high incidence of phenylketonuria in Nagasaki, the westernmost part of Japan, might be due to migration of people with PAH mutations from China and Korea, and geographic factors. PMID: 27173423
- Our study highlighted two novel promoter KLF1 and 3'-region C/EBPalpha motifs in the phenylalanine hydroxylase (PAH) gene which decrease transcription in vitro and, thus, could be considered as PAH expression modifiers. PMID: 27447460
- The results of the in vitro residual PAH activity have major implications, both for our understanding of genotype-phenotype correlations, and thereby existing inconsistencies, but also for the elucidation of the molecular basis of tetrahydrobiopterin (BH4) responsiveness. PMID: 27620137
- Data provide the structural evidence for a dietary I-phenylalanine (Phe) binding pocket at the subunit-subunit interface of a N-terminal regulatory domain (PAH-RD) dimer, and demonstrate that PAH-RD dimerization depends on Phe binding. PMID: 27049649
- The co-expression of two distinct PAH variants revealed possible dominance effects (positive or negative) by one of the variants on residual PAH activity as a result of interallelic complementation. PMID: 26803807
- PAH gene mutation analysis combined with STR linkage analysis can provide rapid and accurate prenatal diagnosis for phenylketonuria families PMID: 26600521
- The mutation spectrum of PAH gene in Henan seems to differ from that of other regions. Independent assortment of mutant alleles may result in a complex genotype-phenotype correlation. PMID: 27264808
- This study identified one novel PAH variant-c.699C>G-and and tries to show a genotype-phenotype relationship also regarding BH4-responsiveness. PMID: 25894915
- This study is the first report on tested population genetic structure using VNTR alleles at the PAH gene PMID: 26025954
- Aberrant methylation is observed in leukocytes of PAH deficient phenylketonuria patients and is influenced by phenylalanine exposure. PMID: 25990862
- We determined phenylalanine hydroxylase function of 30 frequent homozygous and compound heterozygous genotypes covering 55% of the study population. PMID: 25596310
- Mutational spectrum was presented for PAH gene in PAH deficiency patients from different parts of Mexico. New mutations were described. PMID: 24941924
- mutation spectrum of the gene PAH in the Qinghai population was similar to that in other populations in North China PMID: 26575882
- Combining in silico analysis and molecular dynamics simulations (in total 3 mus) we described the structural impact of the mutations, which allowed us to separate 32 out of 34 mutations between groups A and B. PMID: 25750018
- 15 different mutations of phenylalanine hydroxylase gene were detected in patients with phenylketonuria. PMID: 25863075
- R241C, R408Q and Ex6-96A>G are the most common mutations in PAH in phenylalanine hydroxylase deficiency patients. PMID: 25863076
- 15 different mutations were found in 27 unrelated Kurdish PKU patients. IVS4 + 1G > C (c.441 + 1G > C) and IVS7 - 5 T > C (c.843 - 5 T > C) are novel mutations. PAH mutations differ between the Kermanshah province and other parts of Iran. PMID: 24048906
- We demonstrated the high expression of PAH and a large increase of PAH activity in differenciated liver progenitor cells. PMID: 24825084
- findings suggest that common genetic variations in Phenylalanine hydroxylase are associated with verbal memory in healthy adults. PMID: 23898865
- In this study of the PAH mutation spectrum in the Taiwanese population, 139 alleles were identified including 34 different mutations. PMID: 24401910
- Mutations were detected in the exons and flaking introns of PAH gene of 44 families with classical phenylketonuria. PMID: 25449068
- lipoprotein synthesis in PAH-deficient children, particularly in PKU children, was suppressed in early life. PMID: 24607329
- Two polymorphic variants of PAH appear to be risk factors for NSCL/P, rs7485331 and rs12425434 in a Polish population. PMID: 24606907
- This is probably the first report of identification of a significantly low proportion of missense PAH mutations from PKU families and together with the presence of a high proportion of splice, insertion-deletion, and nonsense mutations. PMID: 24130151
- Analysis of the published data shows similar percentage of the "BH4-responsive" variants of a PAH gene in patients from other countries of Eastern Europe PMID: 24350308
- Twenty phenylalanine hydroxylase gene mutations were discovered. PMID: 24510552
- A total of 98 mutations were detected in 110 phenylalanine hydroxylase alleles. PMID: 24510568
- 125 new mutations were found in exons 6, 7 and 12 of PAH in patients with hyperphenylalaninemia. PMID: 24078561
- Genotype-phenotype correlation of PAH gene mutations in phenylketonuria in a Syrian population. PMID: 23856132
- The observed phenotype is not always consistent with genotype predicting effect in Chinese phenylalanine hydroxylase deficiency patients. PMID: 23932990
- The five most prevalent PAH mutations found in patients were p.R408W, IVS12 + 1G>A, p.R261Q, p.R158Q and IVS2 + 5G>C. PMID: 22526846
- A new model for allosteric regulation of phenylalanine hydroxylase: implications for disease and therapeutics. PMID: 23296088
- Thirteen different mutations were identified in the PAH gene in Lebanese patients with phenylalanine hydroxylase deficiency. PMID: 23220018
- The p.G352fsdelG mutation in the PAH gene does not appear to be prevalent in the Moroccan population and would be responsible for only few cases of phenylketonuria. PMID: 22808937
- PAH exon 11 is vulnerable due to a weak 3' splice site. PMID: 22698810
收起更多
-
相关疾病:Phenylketonuria (PKU); Non-phenylketonuria hyperphenylalaninemia (Non-PKU HPA); Hyperphenylalaninemia (HPA)
-
蛋白家族:Biopterin-dependent aromatic amino acid hydroxylase family
-
数据库链接:
HGNC: 8582
OMIM: 261600
KEGG: hsa:5053
STRING: 9606.ENSP00000448059
UniGene: Hs.560019