Recombinant Human Potassium voltage-gated channel subfamily A member 1 (KCNA1), partial
In Stock-
货号:CSB-EP012005HU
-
规格:¥1344
-
图片:
-
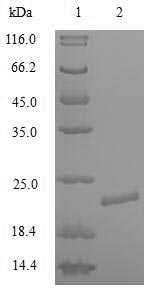 (Tris-Glycine gel) Discontinuous SDS-PAGE (reduced) with 5% enrichment gel and 15% separation gel.
(Tris-Glycine gel) Discontinuous SDS-PAGE (reduced) with 5% enrichment gel and 15% separation gel.
-
-
其他:
产品详情
-
纯度:Greater than 90% as determined by SDS-PAGE.
-
基因名:KCNA1
-
Uniprot No.:
-
别名:AEMK; EA1; Episodic ataxia with myokymia; HBK1; HUK1; Kca1 1; Kcna1; KCNA1_HUMAN; Kcpvd; KV1.1; MBK1; mceph; MGC124402; MGC126782; MGC138385; MK1; MK1, mouse, homolog of KV1.1; Potassium channel protein 1; Potassium voltage gated channel shaker related subfamily member 1; Potassium voltage gated channel subfamily A member 1; Potassium voltage gated channel, shaker related subfamily, member 1 (episodic ataxia with myokymia); Potassium voltage-gated channel subfamily A member 1; RBK1 ; RCK1; Shak; Shaker related subfamily member 1; Voltage gated potassium channel subunit Kv1.1; Voltage-gated K(+) channel HuKI; Voltage-gated potassium channel HBK1; Voltage-gated potassium channel subunit Kv1.1
-
种属:Homo sapiens (Human)
-
蛋白长度:Partial
-
来源:E.coli
-
分子量:22.2kDa
-
表达区域:1-154aa
-
氨基酸序列MTVMSGENVDEASAAPGHPQDGSYPRQADHDDHECCERVVINISGLRFETQLKTLAQFPNTLLGNPKKRMRYFDPLRNEYFFDRNRPSFDAILYYYQSGGRLRRPVNVPLDMFSEEIKFYELGEEAMEKFREDEGFIKEEERPLPEKEYQRQVW
Note: The complete sequence including tag sequence, target protein sequence and linker sequence could be provided upon request. -
蛋白标签:N-terminal 6xHis-tagged
-
产品提供形式:Liquid or Lyophilized powder
Note: We will preferentially ship the format that we have in stock, however, if you have any special requirement for the format, please remark your requirement when placing the order, we will prepare according to your demand. -
缓冲液:Tris-based buffer,50% glycerol
-
储存条件:Store at -20°C/-80°C upon receipt, aliquoting is necessary for mutiple use. Avoid repeated freeze-thaw cycles.
-
保质期:The shelf life is related to many factors, storage state, buffer ingredients, storage temperature and the stability of the protein itself.
Generally, the shelf life of liquid form is 6 months at -20°C/-80°C. The shelf life of lyophilized form is 12 months at -20°C/-80°C. -
货期:3-7 business days
-
注意事项:Repeated freezing and thawing is not recommended. Store working aliquots at 4°C for up to one week.
-
Datasheet & COA:Please contact us to get it.
产品评价

相关产品
靶点详情
-
功能:Voltage-gated potassium channel that mediates transmembrane potassium transport in excitable membranes, primarily in the brain and the central nervous system, but also in the kidney. Contributes to the regulation of the membrane potential and nerve signaling, and prevents neuronal hyperexcitability. Forms tetrameric potassium-selective channels through which potassium ions pass in accordance with their electrochemical gradient. The channel alternates between opened and closed conformations in response to the voltage difference across the membrane. Can form functional homotetrameric channels and heterotetrameric channels that contain variable proportions of KCNA1, KCNA2, KCNA4, KCNA5, KCNA6, KCNA7, and possibly other family members as well; channel properties depend on the type of alpha subunits that are part of the channel. Channel properties are modulated by cytoplasmic beta subunits that regulate the subcellular location of the alpha subunits and promote rapid inactivation of delayed rectifier potassium channels. In vivo, membranes probably contain a mixture of heteromeric potassium channel complexes, making it difficult to assign currents observed in intact tissues to any particular potassium channel family member. Homotetrameric KCNA1 forms a delayed-rectifier potassium channel that opens in response to membrane depolarization, followed by slow spontaneous channel closure. In contrast, a heterotetrameric channel formed by KCNA1 and KCNA4 shows rapid inactivation. Regulates neuronal excitability in hippocampus, especially in mossy fibers and medial perforant path axons, preventing neuronal hyperexcitability. Response to toxins that are selective for KCNA1, respectively for KCNA2, suggests that heteromeric potassium channels composed of both KCNA1 and KCNA2 play a role in pacemaking and regulate the output of deep cerebellar nuclear neurons. May function as down-stream effector for G protein-coupled receptors and inhibit GABAergic inputs to basolateral amygdala neurons. May contribute to the regulation of neurotransmitter release, such as gamma-aminobutyric acid (GABA) release. Plays a role in regulating the generation of action potentials and preventing hyperexcitability in myelinated axons of the vagus nerve, and thereby contributes to the regulation of heart contraction. Required for normal neuromuscular responses. Regulates the frequency of neuronal action potential firing in response to mechanical stimuli, and plays a role in the perception of pain caused by mechanical stimuli, but does not play a role in the perception of pain due to heat stimuli. Required for normal responses to auditory stimuli and precise location of sound sources, but not for sound perception. The use of toxins that block specific channels suggest that it contributes to the regulation of the axonal release of the neurotransmitter dopamine. Required for normal postnatal brain development and normal proliferation of neuronal precursor cells in the brain. Plays a role in the reabsorption of Mg(2+) in the distal convoluted tubules in the kidney and in magnesium ion homeostasis, probably via its effect on the membrane potential.
-
基因功能参考文献:
- In this work, we showed that although both Kv1.1 and Kv1.3 channels are expressed in U87 (glioblastoma), MDA-MB-231 (breast cancer) and LS174 (colon adenocarcinoma) cells, these respond differently to KAaH1 or KAaH2, two homologous Kv1 blockers from scorpion venom PMID: 29415410
- A novel Kv1.1 mutation E283K is associated with a broader EA1 phenotype. Mutant channels show slower activation and positively shifted voltage dependence. PMID: 28666963
- Study reports a novel KCNA1 mutation associated with an episodic ataxia type 1 phenotype and a possible association with malignant hyperthermia (MH). The current report broadens the phenotypes associated with KCNA1 mutations to include possible susceptibility to MH. PMID: 27271339
- Mutation p.Arg324Thr in the KCNA1 gene is pathogenic and results in episodic ataxia type 1 through a dominant-negative effect. PMID: 27477325
- Pharmacogenetic and case-control study evaluated the role of the variants of KCNA1, KCNA2, and KCNV2 in the susceptibility and drug resistance of genetic generalized epilepsies and revealed no significant association between 8 variants of KCNA1, KCNA2, and KCNV2 genes and risk or drug resistance of genetic generalized epilepsies after a Bonferroni correction for multiple comparisons. PMID: 28658141
- we demonstrate that the pathophysiological impact of the I262T mutation entails altered channel gating and defective protein biosynthesis, both of which raise imperative questions that call for further elucidation of the structural and functional roles of the S3 transmembrane segment in Kv1.1 channels. PMID: 26778656
- Herein, we critically evaluate the molecular and biophysical characteristics of the KV1.1 protein in comparison with others and discuss their role in the greater penetrance of KCNA1 mutations in humans leading to the neurological signs of episodic ataxia type 1 PMID: 26825872
- KCNA1 mutations should be considered in patients of all ages with episodic neurological phenotypes, even when ataxia is not present. PMID: 26395884
- These findings provide evidence of an intrinsic cardiac role of Kv1.1 channels and indicate that they may contribute to atrial repolarization and atrial fibrillation susceptibility. PMID: 26162324
- Fine-tuning of Kv1.1 surface expression by RNA editing might contribute to the complexity of neuronal Kv channel regulation. PMID: 25100718
- Novel mutations in KCNA1 genes are associated with episodic ataxia type 1. PMID: 24275721
- Using mutagenesis and analysis of gating currents from gating pore mutations in the Shaker Kv channel, we identified statistically highly significant correlations between VSD function and physicochemical properties of gating pore residues. PMID: 24782544
- The combination of copy number variant and SNPs in KCNA1 (and SCN1A) genes increased the risk for both epilepsy and premature death. PMID: 24372310
- New mutations (R167M, C185W and I407M) were identified in three out of the four families. When expressed in human embryonic kidney cells, all three new mutations resulted in a loss of K(v)1.1 channel function. PMID: 23349320
- characterization of mutations in the potassium channel Kv1.1 PMID: 22609616
- NRG1 increased the intrinsic excitability of FS-PV interneurons which was mediated by increasing the near-threshold responsiveness and decreasing the voltage threshold for action potentials through Kv1.1 PMID: 22158511
- This study suggested that kcna1 missense mutation have been related to Episodic ataxias 1. PMID: 21827920
- Editing of K(V)1.1 channel mRNAs disrupts binding of the N-terminus tip at the intracellular cavity. PMID: 21847110
- Data suggest that episodic ataxia type 1 mutations affect fast inactivation of Kv1.1/1.4 channels by a reduction in either subunit surface expression or altered affinity for the inactivation domain. PMID: 21307345
- Kv1.1 channels are expressed in the beta-cells of several species PMID: 21483673
- integrative genomics approach to a large cohort of medulloblastomas has identified four disparate subgroups (KCNA1)with distinct demographics, clinical presentation, transcriptional profiles, genetic abnormalities, and clinical outcome. PMID: 20823417
- Studies of nerve excitability can identify K(v)1.1 dysfunction in patients with episodic ataxia type 1 PMID: 21106501
- The occurrence of epilepsy in 1 of 2 families with the F414S mutation suggests an interplay of KCNA1 with other genetic factors PMID: 20660867
- An asparagine at position 255 in Kv1.1 is required for normal voltage dependence and kinetics of channel gating. PMID: 19903818
- Variable K(+) channel subunit dysfunction in inherited mutations of KCNA1 PMID: 11773313
- missense mutation involved in episodic ataxia type 1 PMID: 11960817
- I177N, mutation in S1 segment, alters the expression and gating properties of channel expressed in Xenopus oocytes. PMID: 12799903
- Results describe an erbstatin (Erb) analogue as a small molecule inhibitor of the N-type inactivation in potassium channels Kv1.4 and Kv1.1+Kvbeta1. PMID: 15136567
- This study report an unusual family in which the same point mutation in the voltage-gated potassium channel gene KCNA1 resulted in markedly different clinical phenotypes. PMID: 15351427
- coupling between calcium influx and inactivation of voltage-gated A-type K+ channels occurs as a result of membrane depolarization and may contribute to afterhyperpolarization as negative feedback to control neuronal excitability PMID: 15486093
- Results identify palmitoylation as a mechanism for K(+) channel interactions with plasma membrane lipids contributing to electric field-induced conformational alterations. PMID: 15837928
- Myokymia is an autosomal dominant trait caused by mutations in KCNA1, encoding a voltage-gated potassium channel. PMID: 17136396
- This study identified a novel 3-nucleotide deletion mutation in KCNA1 in the episodic ataxia with paroxysmal dyspnea. PMID: 17912752
- The spectrum of neurologic manifestations and neoplasms associated with voltage-gated potassium channel (VGKC) autoimmunity is broader than previously recognized PMID: 18474843
- A novel missense mutation (F414C) KCNA1 identified in an Italian family affected by episodic ataxia type 1. PMID: 18926884
- Contribution of the central hydrophobic residue in the PXP motif of voltage-dependent K+ channels, KCNA1, to S6 flexibility and gating properties. PMID: 19202350
- This study suggested that KCNA1 mutations are associated with a broader clinical phenotype, which may include persistent cerebellar dysfunction and cognitive delay. PMID: 19205071
- studied a family with isolated autosomal dominant hypomagnesemia and used a positional cloning approach to identify an N255D mutation in KCNA1 PMID: 19307729
显示更多
收起更多
-
相关疾病:Episodic ataxia 1 (EA1); Myokymia isolated 1 (MK1)
-
亚细胞定位:Cell membrane; Multi-pass membrane protein. Membrane. Cell projection, axon. Cytoplasmic vesicle. Perikaryon. Endoplasmic reticulum. Cell projection, dendrite. Cell junction. Cell junction, synapse. Cell junction, synapse, presynaptic cell membrane. Cell junction, synapse, presynapse.
-
蛋白家族:Potassium channel family, A (Shaker) (TC 1.A.1.2) subfamily, Kv1.1/KCNA1 sub-subfamily
-
组织特异性:Detected adjacent to nodes of Ranvier in juxtaparanodal zones in spinal cord nerve fibers, but also in paranodal regions in some myelinated spinal cord axons (at protein level). Detected in the islet of Langerhans.
-
数据库链接:
HGNC: 6218
OMIM: 160120
KEGG: hsa:3736
STRING: 9606.ENSP00000371985
UniGene: Hs.416139
Most popular with customers
-
Recombinant Human Semaphorin-4D (SEMA4D), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
Recombinant Dog Angiopoietin-2 (ANGPT2) (Active)
Express system: Mammalian cell
Species: Canis lupus familiaris (Dog) (Canis familiaris)
-
-AC1.jpg)
Recombinant Mouse Claudin-18 (Cldn18)-VLPs (Active)
Express system: Mammalian cell
Species: Mus musculus (Mouse)
-
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
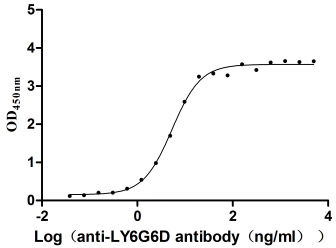
Recombinant Macaca fascicularis lymphocyte antigen 6 family member G6D (LY6G6D) (Active)
Express system: Yeast
Species: Macaca fascicularis (Crab-eating macaque) (Cynomolgus monkey)
-
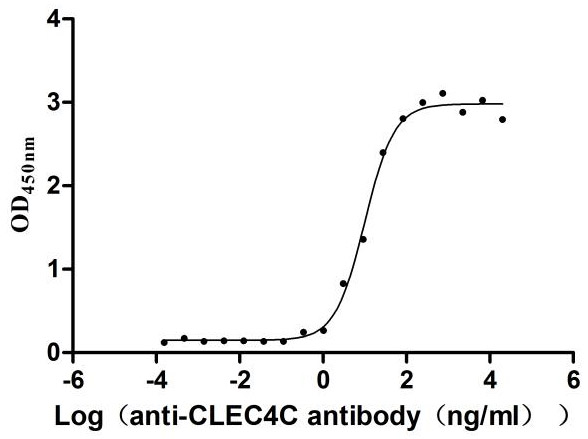
Recombinant Human C-type lectin domain family 4 member C (CLEC4C), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
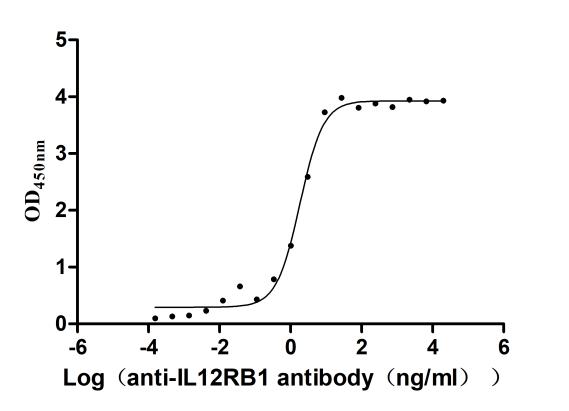
Recombinant Human Interleukin-12 receptor subunit beta-1(IL12RB1),partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
Recombinant DT3C (Diphtheria toxin & spg 3C domain) for Antibody Internalization Assay
Express system: E.coli
Species: Corynephage beta










