OPA1 Antibody
-
货号:CSB-PA016340HA01HU
-
规格:¥440
-
促销:

-
图片:
-
 IHC image of CSB-PA016340HA01HU diluted at 1:300 and staining in paraffin-embedded human small intestine tissue performed on a Leica BondTM system. After dewaxing and hydration, antigen retrieval was mediated by high pressure in a citrate buffer (pH 6.0). Section was blocked with 10% normal goat serum 30min at RT. Then primary antibody (1% BSA) was incubated at 4°C overnight. The primary is detected by a biotinylated secondary antibody and visualized using an HRP conjugated SP system.
IHC image of CSB-PA016340HA01HU diluted at 1:300 and staining in paraffin-embedded human small intestine tissue performed on a Leica BondTM system. After dewaxing and hydration, antigen retrieval was mediated by high pressure in a citrate buffer (pH 6.0). Section was blocked with 10% normal goat serum 30min at RT. Then primary antibody (1% BSA) was incubated at 4°C overnight. The primary is detected by a biotinylated secondary antibody and visualized using an HRP conjugated SP system. -
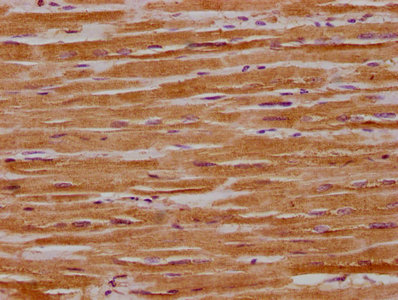 IHC image of CSB-PA016340HA01HU diluted at 1:300 and staining in paraffin-embedded human heart tissue performed on a Leica BondTM system. After dewaxing and hydration, antigen retrieval was mediated by high pressure in a citrate buffer (pH 6.0). Section was blocked with 10% normal goat serum 30min at RT. Then primary antibody (1% BSA) was incubated at 4°C overnight. The primary is detected by a biotinylated secondary antibody and visualized using an HRP conjugated SP system.
IHC image of CSB-PA016340HA01HU diluted at 1:300 and staining in paraffin-embedded human heart tissue performed on a Leica BondTM system. After dewaxing and hydration, antigen retrieval was mediated by high pressure in a citrate buffer (pH 6.0). Section was blocked with 10% normal goat serum 30min at RT. Then primary antibody (1% BSA) was incubated at 4°C overnight. The primary is detected by a biotinylated secondary antibody and visualized using an HRP conjugated SP system.
-
-
其他:
产品详情
-
产品名称:Rabbit anti-Homo sapiens (Human) OPA1 Polyclonal antibody
-
Uniprot No.:O60313
-
基因名:OPA1
-
别名:OPA1; KIAA0567; Dynamin-like 120 kDa protein, mitochondrial; Optic atrophy protein 1
-
宿主:Rabbit
-
反应种属:Human
-
免疫原:Recombinant Human Dynamin-like 120 kDa protein, mitochondrial protein (183-418AA)
-
免疫原种属:Homo sapiens (Human)
-
标记方式:Non-conjugated
本页面中的产品,OPA1 Antibody (CSB-PA016340HA01HU),的标记方式是Non-conjugated。对于OPA1 Antibody,我们还提供其他标记。见下表:
-
克隆类型:Polyclonal
-
抗体亚型:IgG
-
纯化方式:>95%, Protein G purified
-
浓度:It differs from different batches. Please contact us to confirm it.
-
保存缓冲液:Preservative: 0.03% Proclin 300
Constituents: 50% Glycerol, 0.01M PBS, pH 7.4 -
产品提供形式:Liquid
-
应用范围:ELISA, IHC
-
推荐稀释比:
Application Recommended Dilution IHC 1:200-1:500 -
Protocols:
-
储存条件:Upon receipt, store at -20°C or -80°C. Avoid repeated freeze.
-
货期:Basically, we can dispatch the products out in 1-3 working days after receiving your orders. Delivery time maybe differs from different purchasing way or location, please kindly consult your local distributors for specific delivery time.
产品评价

相关产品
靶点详情
-
功能:Dynamin-related GTPase that is essential for normal mitochondrial morphology by regulating the equilibrium between mitochondrial fusion and mitochondrial fission. Coexpression of isoform 1 with shorter alternative products is required for optimal activity in promoting mitochondrial fusion. Binds lipid membranes enriched in negatively charged phospholipids, such as cardiolipin, and promotes membrane tubulation. The intrinsic GTPase activity is low, and is strongly increased by interaction with lipid membranes. Plays a role in remodeling cristae and the release of cytochrome c during apoptosis. Proteolytic processing in response to intrinsic apoptotic signals may lead to disassembly of OPA1 oligomers and release of the caspase activator cytochrome C (CYCS) into the mitochondrial intermembrane space. Plays a role in mitochondrial genome maintenance.; Inactive form produced by cleavage at S1 position by OMA1 following stress conditions that induce loss of mitochondrial membrane potential, leading to negative regulation of mitochondrial fusion.; Isoforms that contain the alternative exon 4b (present in isoform 4 and isoform 5) are required for mitochondrial genome maintenance, possibly by anchoring the mitochondrial nucleoids to the inner mitochondrial membrane.
-
基因功能参考文献:
- Our data indicate that the LEU396ARG mutation in OPA1 is associated with severe dominant optic atrophy. PMID: 29350691
- OPA1 gene therapy prevents retinal ganglion cell loss in a dominant optic atrophy mouse model. PMID: 29410463
- We have generated a human iPSC line IISHDOi003-A from fibroblasts of a patient with a dominant optic atrophy 'plus' phenotype, harbouring a heterozygous mutation, c.1635C>A; p.Ser545Arg, in the OPA1 gene. PMID: 29034899
- OPA1 is a dynamin-related GTPase that controls mitochondrial dynamics, cristae integrity, energetics and mitochondrial DNA maintenance, with eight isoforms being characterized. (Review) PMID: 29382469
- Reports an assessment of the afferent visual system and OCT examination in an Italian cohort of fifty-two fully penetrant probands affected by Autosomal Dominant Optic Atrophy (ADOA) with OPA1 mutations and eight asymptomatic carriers of OPA1 mutations. Visual acuity and OCT data in missense mutations were compared with those associated with mutations inducing haploinsufficiency, and correlated with age in both groups. PMID: 29111013
- we propose that the SIRT4-OPA1 axis is causally linked to mitochondrial dysfunction and altered mitochondrial dynamics that translates into aging-associated decreased mitophagy based on an unbalanced mitochondrial fusion/fission cycle. PMID: 29081403
- Data indicate genotype-phenotype correlations between various types of optic Atrophy 1 (OPA1) mutation and mitophagy. PMID: 28378518
- The results show that a metabolic shift from glycolysis in young to mitochondrial respiration in old normal human fibroblasts occurs during chronological lifespan, and MFN1 and OPA1 regulate this process. PMID: 28758339
- Genetic testing identified disease-causing mutations in 34% of referred cases, with the majority of these in OPA1. Patients with mutations in OPA1 were more likely to have a family history of disease; however, 30.4% of patients without a family history were also found to have an OPA1 mutation. PMID: 28848318
- OPA1 gene mutations were identified in Han Chinese patients with suspected Optic Neuropathy. PMID: 26867657
- Identification of genomic rearrangements or pathogenic variants of OPA1 is meaningful for disease prognosis and proper genetic counseling in DOA consultation. PMID: 28668999
- In brown adipocytes indirect evidence support the notion that OPA1 regulation of fission serves to increase thermogenesis, which thereby contributes to dissipation of energy. PMID: 28427098
- Stabilization of OPA1 impedes cristae remodeling. PMID: 28228254
- Our combination of proteomics, biochemistry, genetics, and electron tomography provides a unifying model for mammalian cristae biogenesis by OPA1 and MICOS. PMID: 27974214
- The splice site mutation (c.985G>T) identified in the present study led to exon 10 skipping (c.985_1065del, p.V329_D355del), suggesting loss-of-function of the GTPase domain of the OPA1 protein, which is likely to cause haplo-insufficiency, a major disease mechanism in DOA. PMID: 26854526
- study identifies a novel pathogenic OPA1 mutation and shows that it is located in the transcript region not prone for NMD activation PMID: 28841713
- OPA1 gene screening in patients with bilateral optic atrophy is an important part of clinical evaluation as it may establish correct clinical diagnosis PMID: 27860320
- OPA1 and cardiolipin cooperate in heterotypic mitochondrial inner membrane fusion. PMID: 28628083
- We propose that OPA1 stabilizes respiratory chain supercomplexes in a conformation that enables respiring mitochondria to compensate a drop in Deltapsim by an explosive matrix pH flash. PMID: 28174208
- We report the first cases of genetically confirmed OPA1-related autosomal-dominant optic atrophy from Singapore, including a novel mutation causing 'ADOA plus' syndrome. PMID: 27858935
- contrary to conventional notion, S-OPA1 is fully competent for maintaining mitochondrial energetics and cristae structure PMID: 28298442
- we analyzed ophthalmological data of a multicentre OPA1 patient cohort and found that women undergo more severe visual loss at adolescence and greater progressive thinning of the retinal nerve fibres than males. Thus, we disclosed a gender-dependent effect on ADOA severity, involving for the first time steroids and Muller glial cells, responsible for RGC degeneration. PMID: 27260406
- The architecture of dendritic arborization in patients with OPA1 mutations is not known, but our data support the idea that loss of dendritic arborization may be involved in the pathogenesis of DOA rather than just population loss. PMID: 28125838
- OPA1 disclosed a de novo heterozygous deletion c.2012+4_2012+7delAGTA resulting in exon 18 and 19 skipping, which was not detected in healthy family members. PMID: 28245802
- This study demonstrated increased mitophagy and excessive mitochondrial fragmentation in primary human cultures associated with DOA plus due to biallelic OPA1 mutations. PMID: 27974645
- The present study identified novel compound heterozygous OPA1 mutations in a patient with recessive optic atrophy, sensorimotor neuropathy and congenital cataracts, indicating an expansion of the clinical spectrum of pathologies associated with OPA1 mutations. PMID: 27150940
- Optic atrophy type 1, caused by mutations in the OPA1 gene is believed to be the most common hereditary optic neuropathy, and most patients inherit a mutation from an affected parent. In this study we used whole-exome sequencing to investigate the genetic aetiology in a patient affected with isolated optic atrophy. Exome results identified a novel de novo OPA1 mutation. PMID: 27265430
- Findings show a new mode of regulation of the mitochondrial fusion proteins, Mfns degradation or OPA1 processing, in response to mitochondrial morphology. PMID: 26935475
- Loss of OPA1 protein function by pathogenic OPA1 gene mutation induces increased mitochondrial fragmentation that promotes instability of the mitochondrial respiratory chain complexes. PMID: 27585216
- Two heterozygous mutations, p.T414P (c.1240A>C) and p.T540P (c.1618A>C), located in the GTPase and middle domains of OPA1, respectively, were identified in two patients.These two different conformational changes might result in decreased GTPase activities that trigger autosomal dominant optic atrophy associated with auditory neuropathy spectrum disorder PMID: 26905822
- A causal link between a pathogenic homozygous OPA1 mutation and hypertrophic cardiomyopathy with optic atrophy was established.It emphasise the vital role played by OPA1 in mitochondrial biogenesis and mtDNA maintenance. PMID: 26561570
- OPA1 variants confer risk of leprosy in Chinese Han population and may affect OPA1 expression, mitochondrial function and antimicrobial pathways. PMID: 26360011
- Genotype-phenotype heterogeneity in OPA1 autosomal-dominant optic atrophy (ADOA). is evident when inner retinal atrophy is examined as a function of age. PMID: 26385429
- Heterozygous mutation in OPA1 disrupts the GTPase domain of OPA1 and is associated with phenotypically variable ADOA Plus. PMID: 26194196
- Identification of copy number variation in the gene for autosomal dominant optic atrophy, OPA1, in a Chinese pedigree PMID: 26400325
- Data show that OPA1 physiological levels are important in cardiovascular health by maintaining mitochondrial shape and respiratory function, while its down-regulation is associated with cardiovascular disease. [review] PMID: 25557256
- increased percentage of apoptotic cells in autosomal dominant optic atrophy patients compared to controls; suggests susceptibility of ADOA cells to oxidative stress and correlation between OPA1 protein dysfunctions and morphological-functional alterations to mitochondria; also results imply sensitivity of mutated protein to free radical damage PMID: 25796301
- Distributed abnormalities of diffusivity indexes might reflect abnormal intracellular mitochondrial morphology as well as alteration of protein levels due to OPA1 mutations. PMID: 25794858
- A recurrent deletion mutation in OPA1 causes autosomal dominant optic atrophy in a Chinese family. PMID: 25374051
- Two heterozygous OPA1 missense mutations affecting highly conserved amino acid positions (p.G488R, p.A495V) were associated with chronic progressive external ophthalmoplegia, parkinsonism, and dementia in two Italian families. PMID: 25820230
- OPA1 mutations induced mitochondrial fragmentation, uncoupled mitochondrial respiration, and elicited dysfunctional bioenergetics. PMID: 25744979
- The results of this study indicated that underlying the hearing impairment in patients carrying OPA1 missense mutations is a disordered synchrony in auditory nerve fibre activity resulting from neural degeneration affecting the terminal dendrites. PMID: 25564500
- Cleavage of the inner membrane fusion factor L-OPA1 is prevented due to the failure to activate the inner membrane protease OMA1 in mitochondria that have a collapsed membrane potential. PMID: 24634514
- a novel way in which OPA1 senses energy substrate availability, which modulates its function in the regulation of mitochondrial architecture in a SLC25A protein-dependent manner. PMID: 25298396
- OMA1 processing is positively correlated with OPA1 cleavage at the S1 site and the regulation of mitochondrial morphology. PMID: 24719224
- The LHON-mtDNA mutations are the most common genetic defects, followed by the OPA1 mutations, in this Chinese cohort. PMID: 25205859
- These findings demonstrate that (a) p53 and Oma1 mediate L-Opa1 processing, (b) mitochondrial fragmentation is involved in CDDP-induced apoptosis in OVCA and CECA cells, and (c) dysregulated mitochondrial dynamics PMID: 25112877
- we report four cases of children affected by Behr syndrome associated with htetrozygous OPA1 mutation PMID: 25012220
- These findings provide additional information regarding the genotype-phenotype correlation and establish the role of the OPA1 gene in Greek patients with autosomal dominant optic atrophy. PMID: 24883014
- patients with mutations in the OPA1 gene has shown that about 20% of them present symptoms of a multiple system disease, which may include peripheral neuropathy [review] PMID: 25137924
显示更多
收起更多
-
相关疾病:Optic atrophy 1 (OPA1); Dominant optic atrophy plus syndrome (DOA+); Behr syndrome (BEHRS); Mitochondrial DNA depletion syndrome 14, cardioencephalomyopathic type (MTDPS14)
-
亚细胞定位:Mitochondrion inner membrane; Single-pass membrane protein. Mitochondrion intermembrane space. Mitochondrion membrane.
-
蛋白家族:TRAFAC class dynamin-like GTPase superfamily, Dynamin/Fzo/YdjA family
-
组织特异性:Highly expressed in retina. Also expressed in brain, testis, heart and skeletal muscle. Isoform 1 expressed in retina, skeletal muscle, heart, lung, ovary, colon, thyroid gland, leukocytes and fetal brain. Isoform 2 expressed in colon, liver, kidney, thyr
-
数据库链接:
HGNC: 8140
OMIM: 125250
KEGG: hsa:4976
STRING: 9606.ENSP00000354681
UniGene: Hs.594504
Most popular with customers
-
-
YWHAB Recombinant Monoclonal Antibody
Applications: ELISA, WB, IF, FC
Species Reactivity: Human, Mouse, Rat
-
Phospho-YAP1 (S127) Recombinant Monoclonal Antibody
Applications: ELISA, WB, IHC
Species Reactivity: Human
-
-
-
-
-