KCNT1 Antibody
-
货号:CSB-PA040093
-
规格:¥880
-
其他:
产品详情
-
Uniprot No.:Q5JUK3
-
基因名:KCNT1
-
别名:bA100C15.2 antibody; EIEE14 antibody; ENFL5 antibody; KCa4.1 antibody; KCNT1 antibody; KCNT1_HUMAN antibody; Potassium channel subfamily T member 1 antibody; Potassium channel, sodium activated subfamily T, member 1 antibody; Potassium channel, subfamily T, member 1 antibody; Sequence like a calcium-activated K+ channel antibody; SLACK antibody; Slo2.2 antibody
-
宿主:Rabbit
-
反应种属:Human,Mouse,Rat
-
免疫原:Synthesized peptide derived from the C-terminal region of Human KCNT1.
-
免疫原种属:Homo sapiens (Human)
-
标记方式:Non-conjugated
-
抗体亚型:IgG
-
纯化方式:The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
-
浓度:It differs from different batches. Please contact us to confirm it.
-
保存缓冲液:Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% sodium azide.
-
产品提供形式:Liquid
-
应用范围:WB, IHC, ELISA
-
推荐稀释比:
Application Recommended Dilution WB 1:500-1:2000 IHC 1:100-1:300 ELISA 1:10000 -
Protocols:
-
储存条件:Upon receipt, store at -20°C or -80°C. Avoid repeated freeze.
-
货期:Basically, we can dispatch the products out in 1-3 working days after receiving your orders. Delivery time maybe differs from different purchasing way or location, please kindly consult your local distributors for specific delivery time.
产品评价

相关产品
靶点详情
-
功能:Outwardly rectifying potassium channel subunit that may coassemble with other Slo-type channel subunits. Activated by high intracellular sodium or chloride levels. Activated upon stimulation of G-protein coupled receptors, such as CHRM1 and GRIA1. May be regulated by calcium in the absence of sodium ions (in vitro).
-
基因功能参考文献:
- G288S missense mutation, associated with seizures and neurodevelopmental delay resulted in larger whole cell K+ currents compared with wild-type KCNT1 currents. PMID: 28747464
- Case report describing 3 infants with malignant migrating partial seizures with KCNT1 mutations accompanied by massive systemic to pulmonary collateral arteries. PMID: 28987752
- Stimulation of Slack K(+) channels alters mass at the plasma membrane by triggering dissociation of Phactr-1. PMID: 27545877
- In the present study, we evaluated two other potential mechanisms for stabilization of Slo2 channels in a closed state: (1) dewetting and collapse of the inner pore (hydrophobic gating) and (2) constriction of the inner pore by tight criss-crossing of the cytoplasmic ends of the S6 alpha-helical segments. PMID: 27682982
- two de novo, heterozygous KCNT1 mutations were identified in two unrelated malignant migrating partial seizures probands. Both mutations induced a marked leftward shift in homomeric channel activation gating. PMID: 26784557
- Better understanding of the mechanisms underlying KCNT1-related disease will produce further improvements in treatment of the associated severe seizure disorders. PMID: 26740507
- The sodium sensitivity of these epilepsy causing mutants probably determines the [Na(+)]i concentration at which these mutants exert their pathological effects. PMID: 26725113
- We demonstrate that KCNT1 mutations are highly pleiotropic and are associated with phenotypes other than nocturnal frontal lobe epilepsy and malignant migrating focal seizures of infancy. PMID: 26122718
- This study demonstrate that KCNT1 mutations are strongly associated with early-onset epileptic encephalopathy. PMID: 26140313
- Five de novo mutations were identified in four genes (SCNN1A, KCNJ16, KCNB2, and KCNT1) in three Brugada syndrome patients (20%) PMID: 25339316
- Nine different mutations of the KCNT1 (Slack) Na(+)-activated K(+) channel give rise to three distinct forms of epilepsy. PMID: 25482562
- Slick channels, in contrast to the similar Slack channels, are the only high-conductance K+ channels strongly sensitive to small changes in cell volume. PMID: 25347289
- Genetic studies reveal two novel genes for Ohtahara Syndrome: KCNT1 and PIGQ. PMID: 24463883
- Novel variations in KCNT1 do not allow prediction of functional phenotypes that might explain, at least in part, the symptoms of malignant migrating partial seizures of infancy (MMPSI). PMID: 24315024
- This gene-wide tagging study revealed no association between KCNT1 17 common variations and susceptibility of GGEs or AEDs (anti-epileptic drugs) efficacy of genetic generalized epilepsies in Chinese population. PMID: 24279416
- This study demonistrated that KCNT1 mutations implicated in epilepsy cause a marked increase in function PMID: 24591078
- this study performed analysis of KCNT1 in two unrelated patients with malignant migrating partial seizures in infancy.Because the G-to-A transition was located at CG dinucleotide sequences as previously reported for KCNT1 mutations, the recurrent occurrence of de novo KCNT1 mutations indicated the hot spots of these locations. PMID: 24029078
- Mutations in KCNT1 cause a severe form of ADNFLE and sporadic NFLE. PMID: 23086396
- Our data identify KCNT1 as a major disease-associated gene in Malignant migrating partial seizures of infancy . PMID: 23086397
显示更多
收起更多
-
相关疾病:Epileptic encephalopathy, early infantile, 14 (EIEE14); Epilepsy, nocturnal frontal lobe, 5 (ENFL5)
-
亚细胞定位:Cell membrane; Multi-pass membrane protein.
-
蛋白家族:Potassium channel family, Calcium-activated (TC 1.A.1.3) subfamily, KCa4.1/KCNT1 sub-subfamily
-
组织特异性:Highest expression in liver, brain and spinal cord. Lowest expression in skeletal muscle.
-
数据库链接:
HGNC: 18865
OMIM: 608167
KEGG: hsa:57582
STRING: 9606.ENSP00000360822
UniGene: Hs.104950
Most popular with customers
-
-
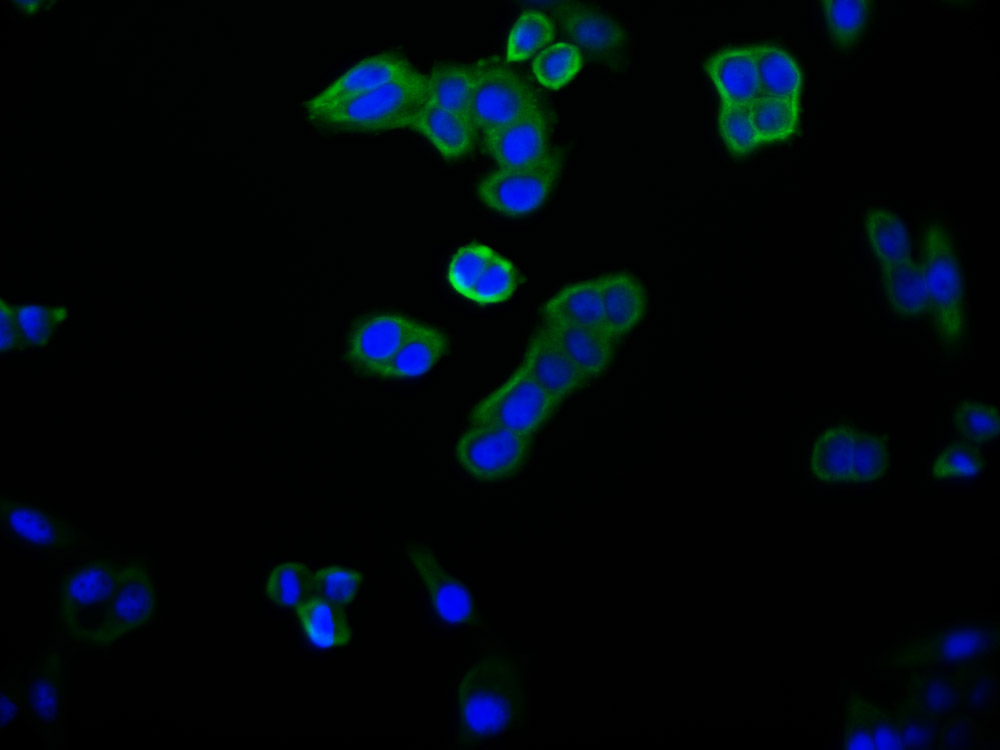
YWHAB Recombinant Monoclonal Antibody
Applications: ELISA, WB, IF, FC
Species Reactivity: Human, Mouse, Rat
-
Phospho-YAP1 (S127) Recombinant Monoclonal Antibody
Applications: ELISA, WB, IHC
Species Reactivity: Human
-
-
-
-
-