GCDH Antibody
-
货号:CSB-PA849798LA01HU
-
规格:¥440
-
促销:

-
图片:
-
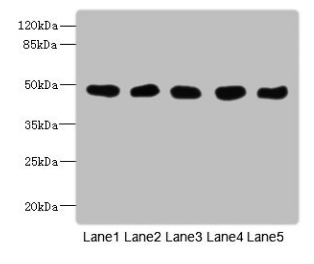 Western Blot
Western Blot
All lanes: GCDH antibody at 8μg/ml
Lane 1: Mouse kidney tissue
Lane 2: Mouse liver tissue
Lane 3: Hela whole cell lysate
Lane 4: MCF-7 whole cell lysate
Lane 5: LO2 whole cell lysate
Secondary
Goat polyclonal to rabbit IgG at 1/10000 dilution
Predicted band size: 49, 48 kDa
Observed band size: 49 kDa -
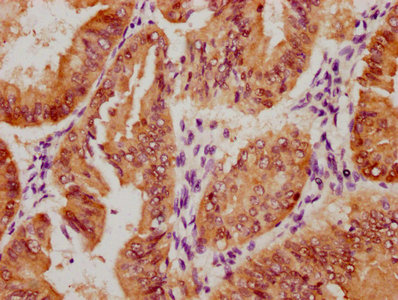
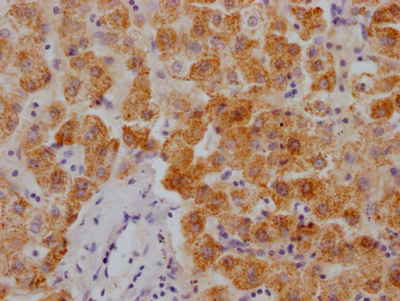
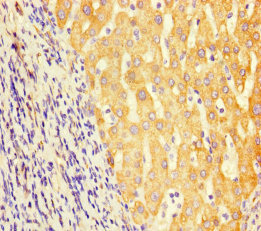 Immunohistochemistry of paraffin-embedded human liver cancer using CSB-PA849798LA01HU at dilution of 1:100
Immunohistochemistry of paraffin-embedded human liver cancer using CSB-PA849798LA01HU at dilution of 1:100 -
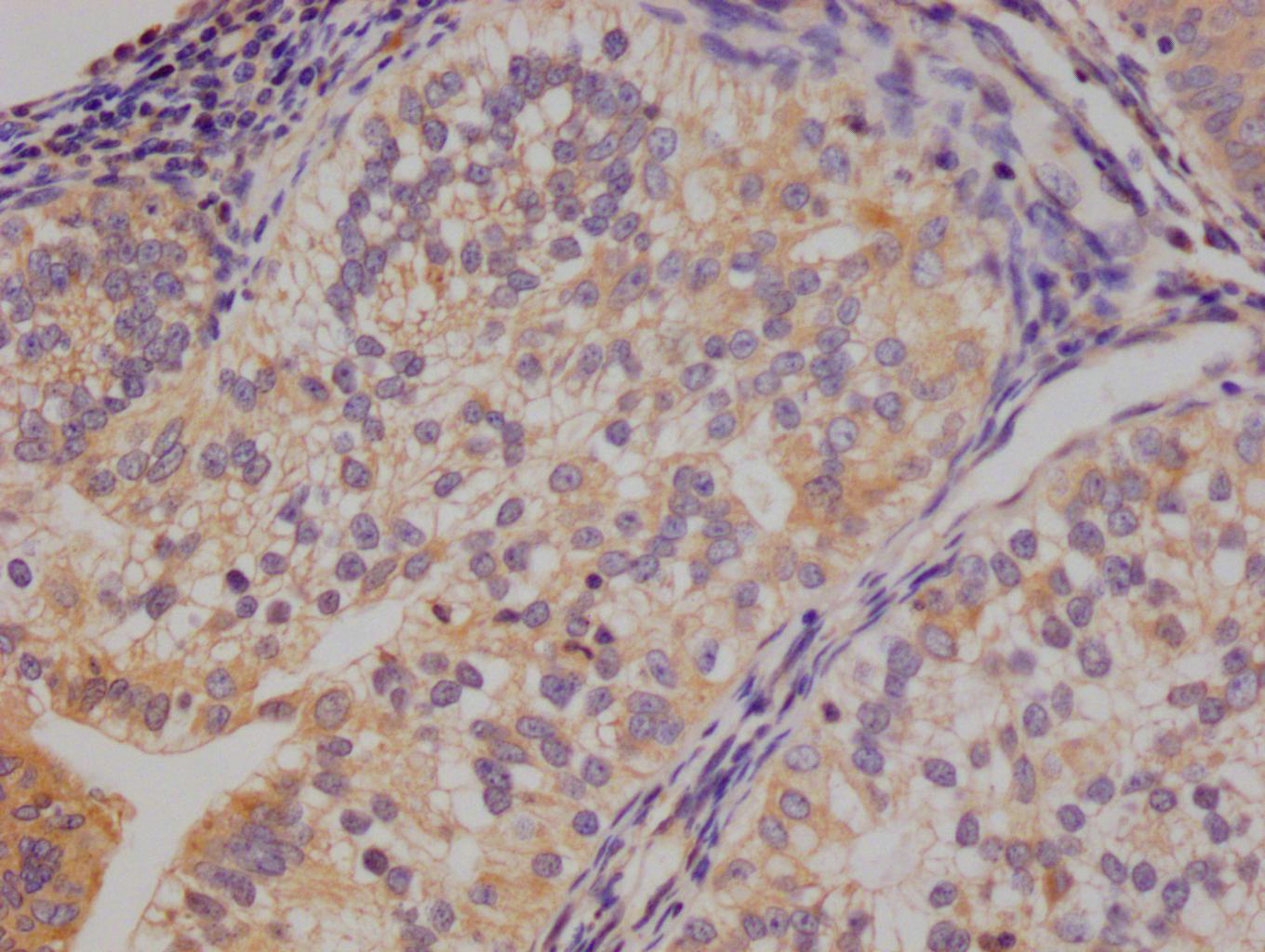
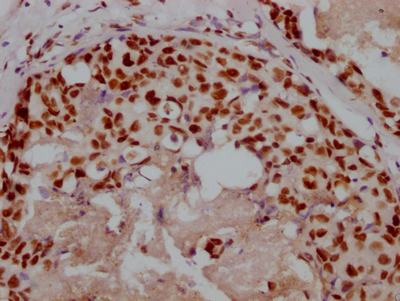
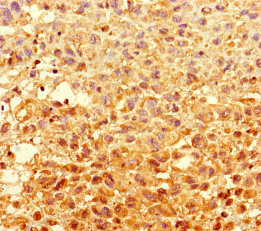 Immunohistochemistry of paraffin-embedded human melanoma using CSB-PA849798LA01HU at dilution of 1:100
Immunohistochemistry of paraffin-embedded human melanoma using CSB-PA849798LA01HU at dilution of 1:100 -
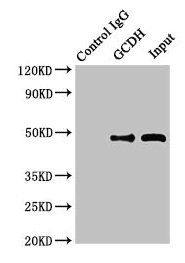 Immunoprecipitating GCDH in Hela whole cell lysate
Immunoprecipitating GCDH in Hela whole cell lysate
Lane 1: Rabbit control IgG instead of (1µg) instead of CSB-PA849798LA01HU in Hela whole cell lysate. For western blotting, a HRP-conjugated Protein G antibody was used as the secondary antibody (1/2000)
Lane 2: CSB-PA849798LA01HU (8µg) + Hela whole cell lysate (500µg)
Lane 3: Hela whole cell lysate (10µg)
-
-
其他:
产品详情
-
产品名称:Rabbit anti-Homo sapiens (Human) GCDH Polyclonal antibody
-
Uniprot No.:Q92947
-
基因名:GCDH
-
别名:ACAD5 antibody; EC 1.3.99.7 antibody; GCD antibody; Gcdh antibody; GCDH_HUMAN antibody; Glutaryl CoA dehydrogenase antibody; Glutaryl CoA dehydrogenase; mitochondrial antibody; Glutaryl Coenzyme A dehydrogenase antibody; Glutaryl-CoA dehydrogenase antibody; mitochondrial antibody; MS781 antibody
-
宿主:Rabbit
-
反应种属:Human, Mouse
-
免疫原:Recombinant Human Glutaryl-CoA dehydrogenase, mitochondrial protein (45-300AA)
-
免疫原种属:Homo sapiens (Human)
-
标记方式:Non-conjugated
本页面中的产品,GCDH Antibody (CSB-PA849798LA01HU),的标记方式是Non-conjugated。对于GCDH Antibody,我们还提供其他标记。见下表:
-
克隆类型:Polyclonal
-
抗体亚型:IgG
-
纯化方式:>95%, Protein G purified
-
浓度:It differs from different batches. Please contact us to confirm it.
-
保存缓冲液:Preservative: 0.03% Proclin 300
Constituents: 50% Glycerol, 0.01M PBS, PH 7.4 -
产品提供形式:Liquid
-
应用范围:ELISA, WB, IHC, IP
-
推荐稀释比:
Application Recommended Dilution WB 1:500-1:5000 IHC 1:20-1:200 IP 1:200-1:2000 -
Protocols:
-
储存条件:Upon receipt, store at -20°C or -80°C. Avoid repeated freeze.
-
货期:Basically, we can dispatch the products out in 1-3 working days after receiving your orders. Delivery time maybe differs from different purchasing way or location, please kindly consult your local distributors for specific delivery time.
产品评价

相关产品
靶点详情
-
功能:Catalyzes the oxidative decarboxylation of glutaryl-CoA to crotonyl-CoA and CO(2) in the degradative pathway of L-lysine, L-hydroxylysine, and L-tryptophan metabolism. It uses electron transfer flavoprotein as its electron acceptor. Isoform Short is inactive.
-
基因功能参考文献:
- Molecular genetics analysis identified 14 different mutations in the GCDH gene in the 18 patients with Glutaric acidemia I PMID: 28389991
- Four mutations of the glutaryl-CoA dehydrogenase (GCDH) gene were identified among the patients with diagnosis of glutaric acidemia type I (GA-I). PMID: 29419857
- We report the allele frequencies for three known Glutaric aciduria type I low excretors GCDH variants (M405V, V400M and R227P) and note that both the M405V and V400M variants are significantly more common in the population of African ancestry compared to the general population PMID: 27397597
- Our data underscore the impact of GCDH protein interactions mediated by amino acid residues on the surface of GCDH required for proper enzymatic activity PMID: 28062662
- Mutations in GCDH gene observed in the present study indicate genetic heterogeneity in GCDH gene among South Indian population. No definite genotype-phenotype correlations were observed. PMID: 26071121
- 2 novel mutations, p.Glu64Asp and p.Gly268Val, account for majority of disease alleles in Cypriot patients with Glutaric aciduria type I; a founder effect for the p.Glu64Asp and the p.Gly268Val can be suggested based on place of origin of mutation carriers PMID: 24973495
- Point mutation of GCDH gene is associated with glutaric academia type I. PMID: 25863083
- 29 GCDH mutations were identified in 23 glutaric aciduria type 1 patients, including 11 novel mutations PMID: 24332224
- Data indicate a homozygous c.1244-2A> C mutation of the glutaryl-CoA dehydrogenase (GCDH) gene in both patients. PMID: 25297592
- These cells displayed decreased levels of GCDH tetramer. PMID: 22231382
- Identification of GCDH gene mutations in four patients with glutaric academia type I. PMID: 23225040
- A homozygous, disease-segregating mutation (p.Val400Met) was identified in the glutaryl-CoA dehydrogenase (GCDH) gene at chromosome 19p13. PMID: 21912879
- physiological concentrations of flavin adenine dinucleotide resulted in a spectacular enhancement of the thermal stabilities of GCDH and prevented enzymatic activity loss PMID: 21968293
- GCDH gene mutations are identified in 8 patients with glutaric aciduria type I PMID: 21811973
- 12 glutaric aciduria type 1 patients were found homozygous for the same A293T mutation in the glutaryl-CoA dehydrogenase (GCDH) gene. PMID: 20732827
- mutational analysis of glutaryl-CoA dehydrogenase in two patients with glutaric aciduria type 1. PMID: 20514322
- Cerebral toxicity caused by GCDH deficiency may induce a state of arteriolar dilation and increased cerebral blood volume. PMID: 20032085
- Three-dimensional structures of human GCD in uncomplexed form and in complex with 4-nitrobutyryl-CoA are reported, and the structural bases for the mechanisms of the dehydrogenation and decarboxylation reactions are proposed. PMID: 15274622
- The s report two GCDH-deficient patients with macrocephaly presenting with progressive neurologic deterioration and a severe leukoencephalopathy during adolescence or adulthood. PMID: 15985591
- An autosomal recessive disease thsat leads to an accumulation of glutaric and 3-hydroxyglutaric acids and secondary carnitine deficiency, (review) PMID: 16368216
- The major rate-determining step in the steady-state turnover of glutaryl-CoA dehydrogenase (GCD) occurs at the release of crotonyl-CoA product; the chemical steps and reoxidation of reduced FAD are much faster than the turnover of wild-type GCD. PMID: 17176108
- GA I is caused by mutations in the GCDH gene, encoding glutaryl-CoA dehydrogenase. PMID: 17661081
- Expression studies of four missense mutations in GCDH indicate that both enzyme instability and impaired enzyme function can underlie the autosomal recessive neurometabolic disorder glutaric aciduria type I. PMID: 18775954
- In the oxidative decarboxylation of glutaryl-coenzyme A (CoA) that is catalyzed by glutaryl-CoA dehydrogenase, glutaconyl-CoA is the presumed enzyme-bound intermediate. PMID: 11705404
显示更多
收起更多
-
相关疾病:Glutaric aciduria 1 (GA1)
-
亚细胞定位:Mitochondrion matrix.
-
蛋白家族:Acyl-CoA dehydrogenase family
-
组织特异性:Isoform Long and isoform Short are expressed in fibroblasts and liver.
-
数据库链接:
HGNC: 4189
OMIM: 231670
KEGG: hsa:2639
STRING: 9606.ENSP00000222214
UniGene: Hs.532699
Most popular with customers
-
-
YWHAB Recombinant Monoclonal Antibody
Applications: ELISA, WB, IF, FC
Species Reactivity: Human, Mouse, Rat
-
Phospho-YAP1 (S127) Recombinant Monoclonal Antibody
Applications: ELISA, WB, IHC
Species Reactivity: Human
-
-
-
-
-