EPM2A Antibody
-
货号:CSB-PA007738ESR1HU
-
规格:¥440
-
促销:

-
图片:
-
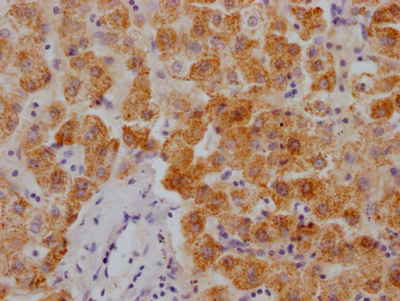
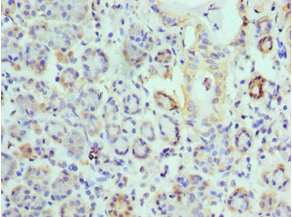 Immunohistochemistry of paraffin-embedded human salivary gland tissue using CSB-PA007738ESR1HU at dilution of 1:100
Immunohistochemistry of paraffin-embedded human salivary gland tissue using CSB-PA007738ESR1HU at dilution of 1:100 -
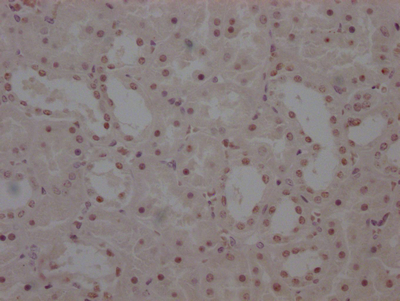
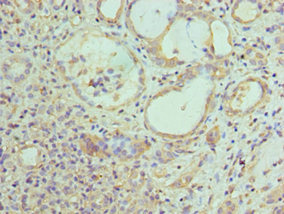 Immunohistochemistry of paraffin-embedded human kidney tissue using CSB-PA007738ESR1HU at dilution of 1:100
Immunohistochemistry of paraffin-embedded human kidney tissue using CSB-PA007738ESR1HU at dilution of 1:100
-
-
其他:
产品详情
-
产品名称:Rabbit anti-Homo sapiens (Human) EPM2A Polyclonal antibody
-
Uniprot No.:O95278
-
基因名:EPM2A
-
别名:Epilepsy progressive myoclonus type 2 Lafora disease (laforin) antibody; Epilepsy progressive myoclonus type 2A Lafora disease (laforin) antibody; EPM2 antibody; Epm2a antibody; Epm2a gene antibody; EPM2A_HUMAN antibody; Lafora PTPase antibody; Laforin antibody; LAFPTPase antibody; LD antibody; LDE antibody; MELF antibody; RP3-466P17.2 antibody
-
宿主:Rabbit
-
反应种属:Human
-
免疫原:Recombinant Human Laforin protein (244-331AA)
-
免疫原种属:Homo sapiens (Human)
-
标记方式:Non-conjugated
-
克隆类型:Polyclonal
-
抗体亚型:IgG
-
纯化方式:Antigen Affinity Purified
-
浓度:It differs from different batches. Please contact us to confirm it.
-
保存缓冲液:PBS with 0.02% sodium azide, 50% glycerol, pH7.3.
-
产品提供形式:Liquid
-
应用范围:ELISA, IHC
-
推荐稀释比:
Application Recommended Dilution IHC 1:20-1:200 -
Protocols:
-
储存条件:Upon receipt, store at -20°C or -80°C. Avoid repeated freeze.
-
货期:Basically, we can dispatch the products out in 1-3 working days after receiving your orders. Delivery time maybe differs from different purchasing way or location, please kindly consult your local distributors for specific delivery time.
产品评价

相关产品
靶点详情
-
功能:Plays an important role in preventing glycogen hyperphosphorylation and the formation of insoluble aggregates, via its activity as glycogen phosphatase, and by promoting the ubiquitination of proteins involved in glycogen metabolism via its interaction with the E3 ubiquitin ligase NHLRC1/malin. Shows strong phosphatase activity towards complex carbohydrates in vitro, avoiding glycogen hyperphosphorylation which is associated with reduced branching and formation of insoluble aggregates. Dephosphorylates phosphotyrosine and synthetic substrates, such as para-nitrophenylphosphate (pNPP), and has low activity with phosphoserine and phosphothreonine substrates (in vitro). Has been shown to dephosphorylate MAPT. Forms a complex with NHLRC1/malin and HSP70, which suppresses the cellular toxicity of misfolded proteins by promoting their degradation through the ubiquitin-proteasome system (UPS). Acts as a scaffold protein to facilitate PPP1R3C/PTG ubiquitination by NHLRC1/malin. Also promotes proteasome-independent protein degradation through the macroautophagy pathway.; does not bind to glycogen. Lacks phosphatase activity and might function as a dominant-negative regulator for the phosphatase activity of isoform 1 and isoform 7.; has phosphatase activity (in vitro).
-
基因功能参考文献:
- rs702304 and rs2235481 within the EPM2A gene were associated with schizophrenia liability. PMID: 27092952
- Laforin prevents the auto-degradation of malin by presenting itself as a substrate. Malin preferentially degrades the phosphatase-inactive laforin monomer. PMID: 26648032
- Laforin-glycan interactions occur with a favourable enthalpic contribution counter-balanced by an unfavourable entropic contribution. PMID: 26578817
- laforin is responsible for glycogen dephosphorylation during exercise and acts during the cytosolic degradation of glycogen PMID: 26216881
- Lafora disease proteins laforin and malin negatively regulate the HIPK2-p53 cell death pathway. PMID: 26102034
- This study suggest that variations in phenotypes of EPM2A-deficient Lafora disease. PMID: 25246353
- novel molecular determinants in the laforin active site that help decipher the mechanism of glucan phosphatase activity. PMID: 25538239
- The crystal structure of laforin bound to phosphoglucan product, reveals its unique integrated tertiary and quaternary structure. PMID: 25544560
- This study identified some Mild Lafora disease have EPM2A mutation. PMID: 25270369
- Studied the role of conformational changes in human laforin structure due to existing single mutation W32G and prepared double mutation W32G/K87A related to loss of glycogen binding. PMID: 24770803
- Polyglucosan body degradation requires a protein assembly that includes laforin and malin. PMID: 24068615
- This study identified the flexibility of K87A mutated laforin structure, with replacement of acidic amino acid to aliphatic amino acid in functional carbohydrate binding module domain, have more impact in abolishing glycogen binding that favors Lafora disease. PMID: 23904258
- These results suggest that cysteine 329 is specifically involved in the dimerization process of laforin. PMID: 23922729
- A new novel mutation of the EPM2A gene is identified that causes Lafora disease. PMID: 23313408
- Studies indicate that laforin directly dephosphorylates glycogen. PMID: 22364389
- Malin forms a functional complex with laforin. This complex promotes the ubiquitination of proteins involved in glycogen metabolism and misregulation of pathways involved in this process results in Lafora body formation. (Review) PMID: 22815132
- This study identified that EPM2 gene mutations leading to Lafora disease in six Turkish families. PMID: 22047982
- alternative splicing could possibly be one of the mechanisms by which EPM2A may regulate the cellular functions of the proteins it codes for PMID: 22036712
- Genetic analysis showed a novel c.659 T>A mutation on exon 3 of the EPM2A gene, in a patient with Lafora's disease. PMID: 21371719
- laforin monomer is the dominant form of the protein and that it contains phosphatase activity. PMID: 21887368
- Laforin and malin are defective in Lafora disease (LD), a neurodegenerative disorder associated with epileptic seizures PMID: 21652633
- glycogen phosphorylation can be considered a catalytic error and laforin a repair enzyme. PMID: 21930129
- results of the present study suggest that phosphorylation of laforin-Ser(25) by AMPK provides a mechanism to modulate the interaction between laforin and malin PMID: 21728993
- study described several novel mutations of EPM2A and NHLRC1 and brought additional data to genetic epidemiology of Lafora disease (LD); emphasized the high mutation rate in patients with classical LD as well as the high negativity rate of skin biopsy PMID: 20738377
- These results suggest that the modification introduced by the laforin-malin complex could affect the subcellular distribution of AMPK beta subunits. PMID: 20534808
- Laforin regulates autophagy. PMID: 20453062
- Laforin is an active phosphatase; therefore, isoforms targeted to different cellular compartments might dephosphorylate and regulate distinct cellular substrates. PMID: 11883934
- identification of mutations in EPM2A with phenotypes of 22 patients (14 families) and identification of two subsyndromes PMID: 12019207
- The EPM2AIP1 gene was identified and characterized in a screen for laforin-interacting proteins with a human brain cDNA library; the specificity of the interaction was confirmed; subcellular colocalization of laforin and EPM2AIP1 protein was demonstrated PMID: 12782127
- Laforin interacts with HIRPI5. PMID: 12915448
- Up to 20% cases of LD are not genetically linked to chromosome 6. We report two sisters affected from bioptically diagnosed LD but without evidence of EPM2A mutation PMID: 14643920
- encodes a 331 amino acid protein that contains an N-terminal carbohydrate-binding domain (CBD) and a C-terminal dual-specificity phosphatase domain. The CBD of laforin targets the protein to Lafora inclusion bodies. PMID: 14706656
- Six novel mutations were identified, one of which is the first mutation specific to the cytoplasmic laforin isoform, implicating this isoform in disease pathogenesis. PMID: 14722920
- Laforin is a Glycogen-Synthase-Kinase-3 Ser 9 phosphatase, and therefore capable of inactivating GS through GSK3. PMID: 16115820
- Laforin does not dephosphorylate GSK3B (beta) in vitro, but possesses the unique ability to utilize a phosphorylated complex carbohydrate as a substrate and this function may be necessary for the maintenance of normal cellular glycogen. PMID: 16901901
- Inactivation of Epm2a resulted in increased Wnt signaling and tumorigenesis PMID: 16959610
- results demonstrate a critical role of dimerization in Laforin function and suggest an important new dimension in protein phosphatase function and in molecular pathogenesis of Lafora's disease PMID: 16971387
- Defects in laforin may lead to increased levels of misfolded and/or target proteins, which may eventually affect the physiological processes of the neuron, and likely to be the primary trigger in the physiopathology of lafora disease. PMID: 17337485
- study concludse that considerable variability in the age at onset of Lafora disease can occur within families; identical mutations can be associated with the classic adolescent presentation, as well as late-onset cases PMID: 17509003
- Regulation of glycogen synthesis by the laforin-malin complex is modulated by the AMP-activated protein kinase pathway. PMID: 18029386
- Results suggest that the altered subcellular localization of mutant proteins of the EPM2A and NHLRC1 genes could be one of the molecular bases of the Lafora disease phenotype. PMID: 18311786
- EPM2A regulated by alternative splicing plays roles in Lafora progressive myoclonus epilepsy. PMID: 18617530
- Laforin and malin interact with misfolded proteins and promote their degradation through the ubiquitin-proteasome system. PMID: 19036738
- phosphorylation of R5/PTG at Ser-8 by AMPK accelerates its laforin/malin-dependent ubiquitination and subsequent proteasomal degradation, which results in a decrease of its glycogenic activity. PMID: 19171932
- Mutations of EPM2A formed aggregates and elicited endoplasm reticulum stress in neuronal cell. PMID: 19403557
- laforin and malin play a role protecting cells from ER-stress, likely contributing to the elimination of unfolded proteins PMID: 19529779
- Laforin is conserved in all vertebrates and a small class of protists; it is not found in other organisms. Additionally, laforin is a functional equivalent of the plant phosphatase SEX4, and it may function to dephosphorylate complex carbohydrates. PMID: 17646401
- Laforin acts as a scaffold that allows the E3 ubiquitin ligase malin to ubiquitinate protein targeting to glycogen (PTG). These results suggest an additional mechanism, involving laforin and malin, in regulating glycogen metabolism. PMID: 18070875
显示更多
收起更多
-
相关疾病:Epilepsy, progressive myoclonic 2 (EPM2)
-
亚细胞定位:Cytoplasm.; [Isoform 1]: Cytoplasm. Endoplasmic reticulum membrane; Peripheral membrane protein; Cytoplasmic side. Cell membrane.; [Isoform 2]: Cytoplasm. Endoplasmic reticulum membrane; Peripheral membrane protein; Cytoplasmic side. Cell membrane. Nucleus.; [Isoform 4]: Cytoplasm. Nucleus.; [Isoform 5]: Cytoplasm. Nucleus.; [Isoform 7]: Cytoplasm.
-
蛋白家族:Protein-tyrosine phosphatase family
-
组织特异性:Expressed in heart, skeletal muscle, kidney, pancreas and brain. Isoform 4 is also expressed in the placenta.
-
数据库链接:
HGNC: 3413
OMIM: 254780
KEGG: hsa:7957
STRING: 9606.ENSP00000356489
UniGene: Hs.486696
Most popular with customers
-
-
YWHAB Recombinant Monoclonal Antibody
Applications: ELISA, WB, IF, FC
Species Reactivity: Human, Mouse, Rat
-
Phospho-YAP1 (S127) Recombinant Monoclonal Antibody
Applications: ELISA, WB, IHC
Species Reactivity: Human
-
-
-
-
-