AGXT Antibody
-
货号:CSB-PA198331
-
规格:¥1100
-
图片:
-
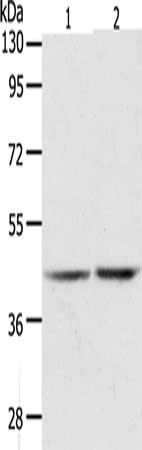 Gel: 10%SDS-PAGE, Lysate: 40 μg, Lane 1-2: Hela cells, human fetal liver tissue, Primary antibody: CSB-PA198331(AGXT Antibody) at dilution 1/400, Secondary antibody: Goat anti rabbit IgG at 1/8000 dilution, Exposure time: 2 minutes
Gel: 10%SDS-PAGE, Lysate: 40 μg, Lane 1-2: Hela cells, human fetal liver tissue, Primary antibody: CSB-PA198331(AGXT Antibody) at dilution 1/400, Secondary antibody: Goat anti rabbit IgG at 1/8000 dilution, Exposure time: 2 minutes
-
-
其他:
产品详情
-
Uniprot No.:P21549
-
基因名:AGXT
-
别名:AGT antibody; AGT1 antibody; Agxt antibody; AGXT1 antibody; Alanine glyoxylate aminotransferase antibody; Alanine glyoxylate aminotransferase3 antibody; Alanine--glyoxylate aminotransferase antibody; EC 2.6.1.44 antibody; EC 2.6.1.51 antibody; Hepatic peroxisomal alanine glyoxylate aminotransferase antibody; Hepatic peroxisomal alanine:glyoxylate aminotransferase antibody; L alanine glyoxylate aminotransferase 1 antibody; MS773 antibody; PH1 antibody; Serine pyruvate aminotransferase antibody; Serine--pyruvate aminotransferase antibody; Serine--pyruvate aminotransferase, mitochondrial antibody; Serine:pyruvate aminotransferase antibody; SPAT antibody; SPT antibody; SPYA_HUMAN antibody; TLH6 antibody
-
宿主:Rabbit
-
反应种属:Human,Mouse,Rat
-
免疫原:Synthetic peptide of Human AGXT
-
免疫原种属:Homo sapiens (Human)
-
标记方式:Non-conjugated
-
抗体亚型:IgG
-
纯化方式:Antigen affinity purification
-
浓度:It differs from different batches. Please contact us to confirm it.
-
保存缓冲液:-20°C, pH7.4 PBS, 0.05% NaN3, 40% Glycerol
-
产品提供形式:Liquid
-
应用范围:ELISA,WB
-
推荐稀释比:
Application Recommended Dilution ELISA 1:1000-1:2000 WB 1:200-1:1000 -
Protocols:
-
储存条件:Upon receipt, store at -20°C or -80°C. Avoid repeated freeze.
-
货期:Basically, we can dispatch the products out in 1-3 working days after receiving your orders. Delivery time maybe differs from different purchasing way or location, please kindly consult your local distributors for specific delivery time.
产品评价

相关产品
靶点详情
-
基因功能参考文献:
- Mutations at the dimer interface of alanine-glyoxylate aminotransferase alter enzyme structure/activity, leading to primary hyperoxaluria type I and determine the cellular response to vitamin B6. PMID: 29110180
- two novel AGXT missense mutations (p.M49L and p.N72I), which will enrich the AGXT mutation database and provide a better comprehension of PH1 pathogenesis, were identified in a big Chinese PH1 family. Significant morphological and structural difference of kidney stones from two siblings with the same genotype observed in this study displays the heterogeneity of genotype-phenotype correlation PMID: 27644547
- AGXT mutational analysis was performed to confirm the diagnosis of PH1. PMID: 28161266
- this study identifies a novel AGXT gene mutation in primary hyperoxaluria after kidney transplantation failure in Tunisian patient PMID: 27568336
- AGXT gene sequencing is now the choice of diagnosis of Primary hyperoxaluria type 1 (PH1)due to its non-invasive nature compared to liver enzyme assay. Early diagnosis and accurate treatment in PH1 is important for better patient outcomes. PMID: 27915025
- Caenorhabditis elegans AGXT-1 is a mitochondrial and temperature-adapted ortholog of peroxisomal human AGT1. PMID: 27179589
- Novel AGXT mutations in a Tunisian population with primary hyperoxaluria type 1. PMID: 27935012
- The novel p.Gln137Hisfs*19 AGXT mutation detected in this study extends the spectrum of known primary hyperoxaluria type I AGXT gene mutations in Tunisia. PMID: 27659337
- Data show that onomeric minor allele of human alanine glyoxylate aminotransferase (AGT-Mi) binds pyridoxal 5-phosphate (PLP) but does not display catalytic activity. PMID: 27720751
- Letter/Case Report: novel missense AGXT gene mutation in a Sri Lankan family with primary hyperoxaluria type 1. PMID: 26693850
- Primary hyperoxaluria type 1 (PH1) is due to a defect in the AGXT gene. The aim of our study was to analyze the mutations causing PH1 in the Moroccan population PMID: 26383609
- In conclusion, this study of an unprecedented number of primary hyperoxaluria type 1 patients showed geno-phenotype associations that have not been previously reported. PMID: 24988064
- The pathogenic mutation G47R causes misfolding of alanine:glyoxylate aminotransferase. PMID: 26149463
- A review of the current knowledge of the biochemical properties of liver peroxisomal alanine:glyoxylate aminotransferase and of the molecular defects caused by single point mutations associated with Primary Hyperoxaluria Type 1. PMID: 25620715
- S81L and G170R mutations of AGT is associated with Primary Hyperoxaluria type I in homozygosis and heterozygosis. PMID: 24990153
- AGT missense mutations associated with Primary Hyperoxaluria Type 1, were characterized. PMID: 24718375
- Data suggest that dequalinium chloride (DECA) may be a pharmacologic strategy to treat primary hyperoxaluria 1 (PH1) patients with mutations in alanine:glyoxylate aminotransferase (AGT). PMID: 25237136
- These are the first cases of primary hyperoxaluria type 1 to be diagnosed by clinical manifestations and AGXT gene mutations in mainland China. PMID: 24934730
- data imply that the AGT Pro11Leu polymorphism is not directly responsible for the low incidence of stone formation in black South Africans. PMID: 24344980
- Modeling of the mutations on a 1.9 A crystal structure suggests that Primary hyperoxaluria type I causing mutants perturb locally the native structure of AGT. PMID: 24205397
- The view presented has important implications for the development of new therapeutic strategies based on targeting specific elements of alanine-glyoxylate aminotransferase homeostasis PMID: 23956997
- Gly161 mutations in alanine:glyoxylate aminotransferase is associated with Primary Hyperoxaluria Type I. PMID: 24055001
- Solved is the X-ray crystal structure of the S187F variant of AGT to a resolution of 2.9 A. PMID: 23589421
- Identification of a double mutation c.32C>T (Pro11Leu) and c.731T>C (p.Ile244Thr) in AGXT gene in five unrelated Tunisian families with primary hyperoxaluria type 1 disease. PMID: 24012869
- Three novel mutations detected in the AGXT gene associated with primary hyperoxaluria type 1 in a Tunisian patient population. PMID: 23810941
- Four of the most common mutations in primary hyperoxaluria type 1 unmask the cryptic mitochondrial targeting sequence of alanine:glyoxylate aminotransferase encoded by the polymorphic minor allele PMID: 23229545
- The molecular mechanism of recognition by the peroxisomal receptor Pex5p, in complex with alanine-glyoxylate aminotransferase revealed by X-ray crystallography. PMID: 22529745
- These results suggest that the N-terminal extension plays an essential role in allowing AGT to attain its correct conformation and functional activity. PMID: 22198249
- A side-by-side comparison was performed between normal AGT and nine purified recombinant pathogenic variants in terms of catalytic activity, coenzyme binding mode and affinity, spectroscopic features, oligomerization, and thermal stability. PMID: 22018727
- selected aspects of the biochemical properties of the two allelic forms of AGXT and of some primary hyperoxaluria type 1-causing variants (review) PMID: 22201765
- Partially unfolded states of AGXT strongly interact with Hsc70 and Hsp90 chaperones. PMID: 21103899
- Selected AGXT gene mutations analysis provides a genetic diagnosis in 28% of Tunisian patients with primary hyperoxaluria. PMID: 21612638
- Data show that P11L mutation is responsible for the urea sensitivity of AGT-Mi. PMID: 20713123
- mutation of the AGXT gene, showing the patient to be compound heterozygous for the c.33_34InsC and c.508G > A mutations PMID: 20020206
- Genotype-phenotype correlation in primary hypoxaluria type 1: the p. Gly170Arg AGXT mutation is associated with a better outcome. PMID: 20016466
- Molecular defects of the glycine 41 variants of alanine glyoxylate aminotransferase associated with primary hyperoxaluria type I. PMID: 20133649
- investigated occurrence of Pro11Leu polymorphism in both herder & agriculturalist populations from Central Asia; findings show distribution of variation observed in the AGXT gene could be due to demographic history, rather than local adaptation to diet PMID: 20059472
- crystal structure of AGT consists of an intimate dimer in which an extended N-terminal segment of 21 amino acids from one subunit wraps as an elongated irregular coil around the outside of the crystallographic symmetry-related subunit. PMID: 20208150
- 3D picture of an in vivo early ATP-dependent step of the folding reaction cycle of the chaperonin and supports a GroEL functional model in which the chaperonin promotes folding of the AGXT-LTM mutant protein through forced unfolding mechanism PMID: 20056599
- serine:pyruvate aminotransferase expression is regulated by Sp1, AP-2 and PKA PMID: 12169688
- crystal structure of normal human AGT complexed to the competitive inhibitor amino-oxyacetic acid to 2.5A PMID: 12899834
- Early detection of Gly170Arg and Phe152Ile mutations in PH1 has important clinical implications because of their association with pyridoxine responsiveness and clinical outcome PMID: 15253729
- report describing 3 AGXT gene mutations in Chinese patients with primary hyperoxaluria type 1 PMID: 15365967
- mutations with serious consequences in vivo may not be inherently catalytically inactive and may be rescuable PMID: 15802217
- human AGT interacts with human Pex5p in mammalian cells, but not yeast cells; type 1 peroxisomal targeting sequence(PTS1)is located entirely within the smaller C-terminal structural domain of 110 amino acids PMID: 15911627
- Determination of crystal structure of AGT has enabled effects of some of most important missense mutations in AGXT gene to be rationalised in terms of AGT folding, dimerization and stability. New possibilities for design of pharmacological agents. PMID: 15961951
- Presentation and role of transplantation [kidney and/or liver] in adult patients with type 1 primary hyperoxaluria and the 1244T AGXT mutation in a university hospital in Spain are presented. PMID: 16912707
- The effects of missense mutations on enzyme activity, dimerization, aggregation, and turnover were investigated. PMID: 16971151
- expressed wild-type human AGT1 was predominantly localized in mouse hepatocellular peroxisomes, whereas the most common mutant form of AGT1 (G170R) was localized predominantly in the mitochondria PMID: 17110443
- AGXT is even more variable than formerly believed in the diagnosis of primary hyperoxaluria PMID: 17460142
显示更多
收起更多
-
相关疾病:Hyperoxaluria primary 1 (HP1)
-
亚细胞定位:Peroxisome. Mitochondrion.
-
蛋白家族:Class-V pyridoxal-phosphate-dependent aminotransferase family
-
组织特异性:Liver.
-
数据库链接:
HGNC: 341
OMIM: 259900
KEGG: hsa:189
STRING: 9606.ENSP00000302620
UniGene: Hs.144567
Most popular with customers
-
-
Phospho-YAP1 (S127) Recombinant Monoclonal Antibody
Applications: ELISA, WB, IHC
Species Reactivity: Human
-
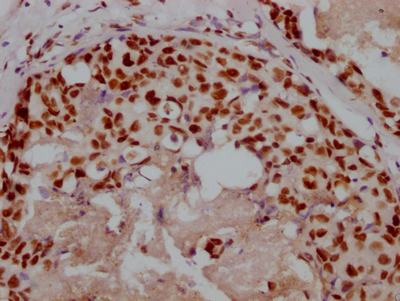
-
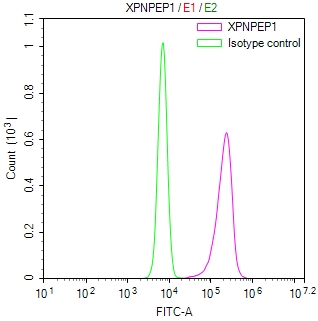
-
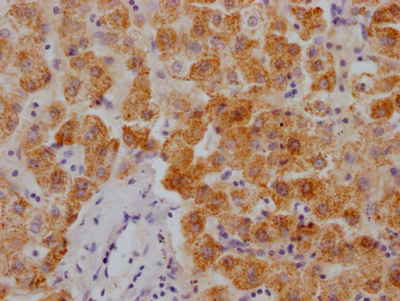
-
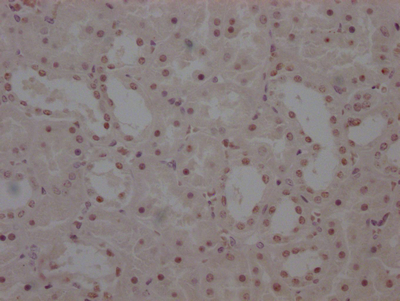
-
-