Recombinant Human Dysferlin (DYSF), partial
-
货号:CSB-YP007314HU
-
规格:
-
来源:Yeast
-
其他:
-
货号:CSB-EP007314HU
-
规格:
-
来源:E.coli
-
其他:
-
货号:CSB-EP007314HU-B
-
规格:
-
来源:E.coli
-
共轭:Avi-tag Biotinylated
E. coli biotin ligase (BirA) is highly specific in covalently attaching biotin to the 15 amino acid AviTag peptide. This recombinant protein was biotinylated in vivo by AviTag-BirA technology, which method is BriA catalyzes amide linkage between the biotin and the specific lysine of the AviTag.
-
其他:
-
货号:CSB-BP007314HU
-
规格:
-
来源:Baculovirus
-
其他:
-
货号:CSB-MP007314HU
-
规格:
-
来源:Mammalian cell
-
其他:
产品详情
-
纯度:>85% (SDS-PAGE)
-
基因名:
-
Uniprot No.:
-
别名:DMAT; DYSF; DYSF_HUMAN; Dysferlin; Dysferlin limb girdle muscular dystrophy 2B (autosomal recessive); Dysferlin limb girdle muscular dystrophy 2B; Dystrophy associated fer 1 like 1; Dystrophy associated fer 1 like protein; Dystrophy associated fer1 like 1; Dystrophy associated fer1 like protein; Dystrophy-associated fer-1-like protein; Fer 1 like protein 1; Fer-1-like protein 1; Fer1 like protein 1; FER1L1; FLJ00175; FLJ90168; LGMD 2B; LGMD2B; Limb girdle muscular dystrophy 2B (autosomal recessive); Limb girdle muscular dystrophy 2B; Miyoshi myopathy; MM; MMD1
-
种属:Homo sapiens (Human)
-
蛋白长度:Partial
-
蛋白标签:Tag type will be determined during the manufacturing process.
The tag type will be determined during production process. If you have specified tag type, please tell us and we will develop the specified tag preferentially. -
产品提供形式:Lyophilized powder
Note: We will preferentially ship the format that we have in stock, however, if you have any special requirement for the format, please remark your requirement when placing the order, we will prepare according to your demand. -
复溶:We recommend that this vial be briefly centrifuged prior to opening to bring the contents to the bottom. Please reconstitute protein in deionized sterile water to a concentration of 0.1-1.0 mg/mL.We recommend to add 5-50% of glycerol (final concentration) and aliquot for long-term storage at -20℃/-80℃. Our default final concentration of glycerol is 50%. Customers could use it as reference.
-
储存条件:Store at -20°C/-80°C upon receipt, aliquoting is necessary for mutiple use. Avoid repeated freeze-thaw cycles.
-
保质期:The shelf life is related to many factors, storage state, buffer ingredients, storage temperature and the stability of the protein itself.
Generally, the shelf life of liquid form is 6 months at -20°C/-80°C. The shelf life of lyophilized form is 12 months at -20°C/-80°C. -
货期:Delivery time may differ from different purchasing way or location, please kindly consult your local distributors for specific delivery time.Note: All of our proteins are default shipped with normal blue ice packs, if you request to ship with dry ice, please communicate with us in advance and extra fees will be charged.
-
注意事项:Repeated freezing and thawing is not recommended. Store working aliquots at 4°C for up to one week.
-
Datasheet :Please contact us to get it.
产品评价

相关产品
靶点详情
-
功能:Key calcium ion sensor involved in the Ca(2+)-triggered synaptic vesicle-plasma membrane fusion. Plays a role in the sarcolemma repair mechanism of both skeletal muscle and cardiomyocytes that permits rapid resealing of membranes disrupted by mechanical stress.
-
基因功能参考文献:
- arginine-rich motif crucial for phosphatidylserine accumulation in sarcolemma repair PMID: 27641898
- A novel duplication of 22 bases (c.897_918dup; p.Gly307Leufs5X) in the DYSF gene was identified in a family suffering from Miyoshi myopathy PMID: 29209666
- This review detailed the different partners and function of dysferlin and positions the sarcolemma repair in normal and pathological conditions. [Review] PMID: 29480214
- Immunofluorescence demonstrated that the percentage of complex I- and complex IV-deficient fibres was higher in patients with DYSF mutations than in age-matched controls. No clonally expanded mtDNA deletions were detected using long-range PCR in any of the analysed muscle fibres. Complex I and complex IV deficiency is higher in patients than age matched controls but patients do not have rearrangements of the mtDNA. PMID: 27666772
- Data suggest that dysferlin exhibits modular architecture of 4 tertiary domains: 1) C2A, readily removed as solo domain; 2) midregion C2B-C2C-Fer-DysF, excised as intact module with several dynamic folding options; 3) C-terminal four-C2 domain module; 4) calpain-2-cleaved mini-dysferlinC72, particularly resistant to proteolysis. Missense variant L344P in muscular dystrophy patient largely escapes proteasomal surveillance. PMID: 28904177
- dysferlin has membrane tubulating capacity and that it shapes the T-tubule system. PMID: 28104817
- Human deltoid muscle biopsies of 5 Chilean dysferlinopathy patients exhibited the presence of muscular connexins (Cx40.1, Cx43 and Cx45). PMID: 27229680
- This review suggested that the functions of dysferlin in vesicle trafficking and membrane remodeling in skeletal muscle. PMID: 27349407
- DYSF expression is significantly upregulated in human masticatory mucosa during wound healing PMID: 28005267
- DYSF mutations in Chinese patients clustered in the N-terminal region of the gene. Exonic rearrangements were found in 23% of patients with only one pathogenic mutation identified by Sanger sequencing or NGS. The novel mutations found in this study greatly expanded the mutational spectrum of dysferlinopathy. PMID: 27647186
- This study showed that 4 patients with Inflammatory Myopathy associated with DYSF mutation. PMID: 26911292
- results support a function for dysferlin as a calcium-sensing SNARE effector for membrane fusion events PMID: 27226605
- These differences in the structural dynamics of the predicted binding site suggest that mutation R959W alters recognition dynamics of the inner DysF domain. PMID: 26806107
- This study demonstrated that novel mutation of DYSF in patient with Dysferlinopathy in Iran. PMID: 26671124
- By targeting DYSF premRNA introns harbouring differentially defined 3' splice sites (3' SS), we found that target introns encoding weakly defined 3' SSs were trans-spliced successfully in vitro in human myoblasts also in vivo in skeletal muscle of mice. PMID: 25904108
- minigene strategy is an efficient tool for the detection of splicing defects in dysferlinopathies, which could allow for a better comprehension of splicing defects due to mutations and could improve prediction tools evaluating splicing defects PMID: 25312915
- Dysferlin carrier frequency and the number of affected individuals at risk for dysferlinopathy could be higher than previously estimated. PMID: 24838345
- Our study underlines clinical heterogeneity and a high proportion of novel mutations for dysferlin in Chinese patients affected with dysferlinopathy. PMID: 25591676
- results provide the mechanism for dysferlin-mediated repair of skeletal muscle sarcolemma and identify ASM as a potential therapy for dysferlinopathy PMID: 24967968
- These novel observations of conspicuous intermyofibrillar lipid and progressive adipocyte replacement in dysferlin-deficient muscles. PMID: 24685690
- Our results suggest that dysferlin protein levels of
- The crystal structure of the human dysferlin inner DysF domain shows that most of the pathogenic mutations are part of aromatic/arginine stacks that hold the domain in a folded conformation. PMID: 24438169
- The tricomplex Fam65b-HDAC6-dysferlin is transient. PMID: 24687993
- all dysferlin domains bind Ca(2+) albeit with varying affinity and stoichiometry PMID: 24461013
- distinct membrane protein signature specific to patients with Diamond-Blackfan Anemia PMID: 24454878
- Alternate splicing of the dysferlin C2A domain confers Ca(2+)-dependent and Ca(2+)-independent binding for membrane repair. PMID: 24239457
- These data suggest that although dysferlin is not an integral part of the dystrophin-glycoprotein complex, its expression is altered in Duchenne muscular dystrophy. PMID: 24902367
- our results identify dysferlin as a newly identified binding partner of AbetaPP PMID: 24091414
- We described 8 Chinese patients with dysferlinopathy PMID: 23254335
- a direct interaction of dysferlin with Trim72/MG53, AHNAK, cytoplasmic dynein, myomesin-2 and calsequestrin-1, but not with caveolin-3 or dystrophin, is reported. PMID: 23792176
- Data indicate that dysferlin, otoferlin, and myoferlin do not merely passively adsorb to membranes but actively sculpt lipid bilayers. PMID: 23859474
- dysferlin is involved in regulating cellular interactions and has a role in inflammatory cells PMID: 23558685
- study reported 4 novel mutations and 2 cases of dysferlinopathy in which patients exhibited a reduction of sarcolemmal dysferlin in conjunction with cytoplasmic retention PMID: 23519732
- Dysferlin is subject to enzymatic cleavage releasing a synaptotagmin-like fragment with a specialized protein- or phospholipid-binding role for muscle membrane repair. PMID: 23516275
- Dysferlin-peptides reallocate mutated dysferlin thereby restoring function PMID: 23185377
- we observed 40 Japanese patients in 36 families with limb girdle muscular dystrophy 2B in whom dysferlin mutations were confirmed PMID: 23243261
- provide proof of principle that AAV5 mediated delivery of dysferlin is a highly promising strategy for treatment of dysferlinopathies and has far-reaching implications for the therapeutic delivery of other large genes PMID: 22720081
- In Koreans with dysferlinopathy, DYSF mutations appeared to cluster in the N-terminal region. PMID: 22297152
- The aim of the study was to determine whether dysferlin expression in peripheral blood monocytes correlates with that in skeletal muscle. PMID: 22194990
- C2 domains mediate high affinity self-association of dysferlin in a parallel homodimer PMID: 22110769
- Studies indicate that dysferlinopathies are autosomal recessive disorders caused by mutations in the dysferlin (DYSF) gene, encoding the dysferlin protein. PMID: 21556485
- these data further support the claims that dysferlin not only mediates membrane repair but also trafficking of client proteins, ultimately, help bridging dysferlinopathies to aberrant membrane signaling. PMID: 22037454
- This study presents the first direct and conclusive evidence that an amount of Dysferlin
- Data suggest dysferlin has an important function in the internal membrane systems of skeletal muscle, involved in calcium homeostasis and excitation-contraction coupling. PMID: 22043020
- A simple and rapid screening method to detect hot spot mutations in the dysferlin gene is essential for the diagnosis of dysferlinopathy. PMID: 21173544
- A novel mutation in exon 47 (c.5289G>C) of the dysferlin gene in the heterozygous state, causing an amino acid change (p.Glu1763Asp), was detected in 2 patients PMID: 21658164
- A new computational method establishes an increase in the mean average prediction precision for dysferlin protein partners, which is important for new targeted therapies. PMID: 21280221
- MG53, annexin A1, and dysferlin localize to the t-tubule network and show enriched labeling at longitudinal tubules of the t-system in overstretch PMID: 21412170
- Dysferlin function in intracellular vesicles and its implication in muscle membrane resealing. PMID: 21119217
- B cell depletion with rituximab/dysferlin monoclonal antibody has been proved useful in the treatment of two patients affected by muscular dystrophy. There may be a possible role for B cells in the immune system involvement of this muscle disorder. PMID: 20618995
显示更多
收起更多
-
相关疾病:Limb-girdle muscular dystrophy 2B (LGMD2B); Miyoshi muscular dystrophy 1 (MMD1); Distal myopathy with anterior tibial onset (DMAT)
-
亚细胞定位:Cell membrane, sarcolemma; Single-pass type II membrane protein. Cytoplasmic vesicle membrane; Single-pass type II membrane protein. Cell membrane.
-
蛋白家族:Ferlin family
-
组织特异性:Expressed in skeletal muscle, myoblast, myotube and in the syncytiotrophoblast (STB) of the placenta (at protein level). Ubiquitous. Highly expressed in skeletal muscle. Also found in heart, brain, spleen, intestine, placenta and at lower levels in liver,
-
数据库链接:
HGNC: 3097
OMIM: 253601
KEGG: hsa:8291
STRING: 9606.ENSP00000386881
UniGene: Hs.252180
Most popular with customers
-
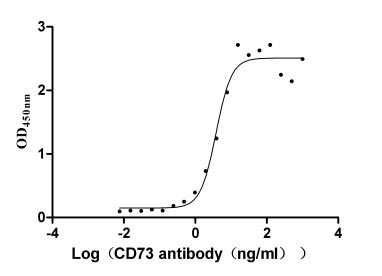
Recombinant Human 5'-nucleotidase (NT5E) (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
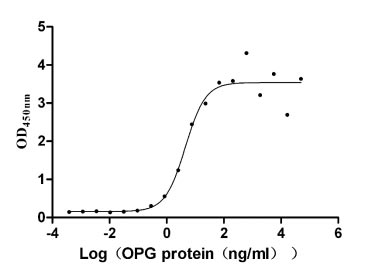
Recombinant Human Tumor necrosis factor receptor superfamily member 11B (TNFRSF11B) (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
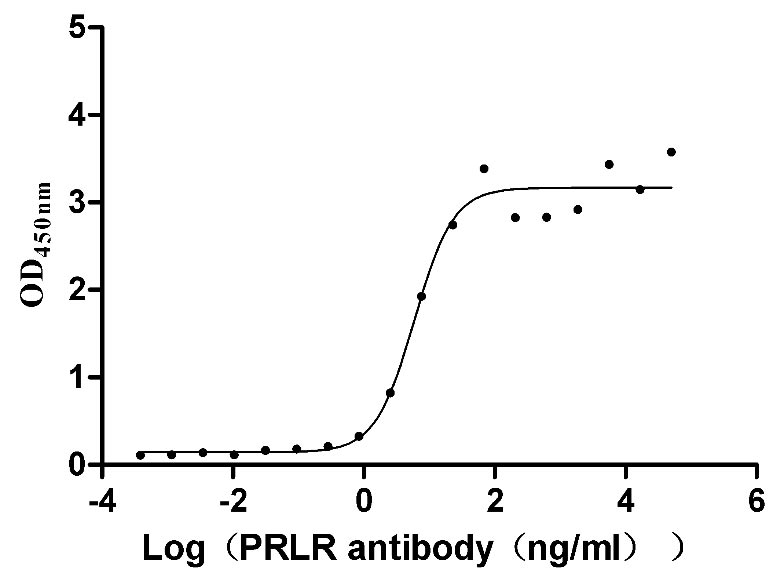
Recombinant Mouse Prolactin receptor (Prlr), partial (Active)
Express system: Mammalian cell
Species: Mus musculus (Mouse)
-
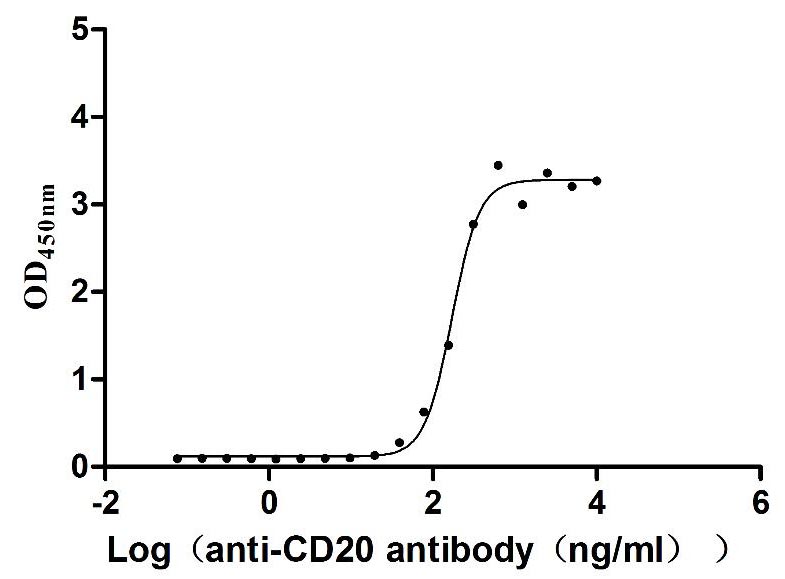
Recombinant Dog B-lymphocyte antigen CD20 (MS4A1)-VLPs (Active)
Express system: Mammalian cell
Species: Canis lupus familiaris (Dog) (Canis familiaris)
-
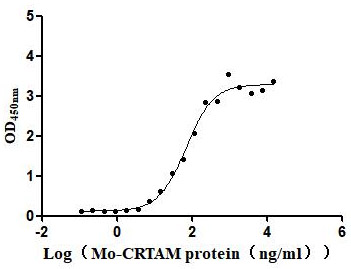
Recombinant Mouse Cell adhesion molecule 1 (Cadm1), partial (Active)
Express system: Mammalian cell
Species: Mus musculus (Mouse)
-

Recombinant Human Myosin regulatory light chain 12A (MYL12A) (Active)
Express system: E.coli
Species: Homo sapiens (Human)
-
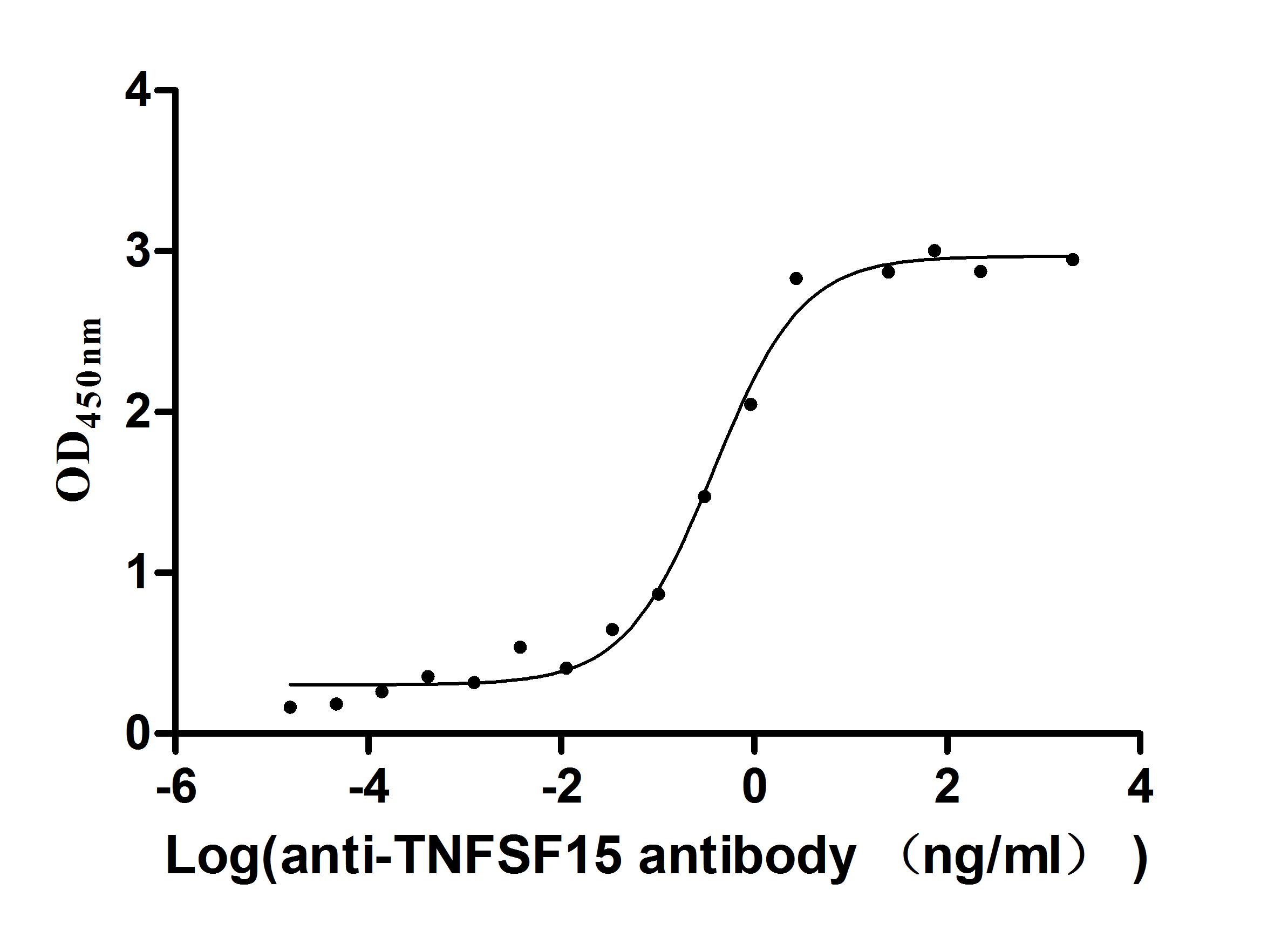
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
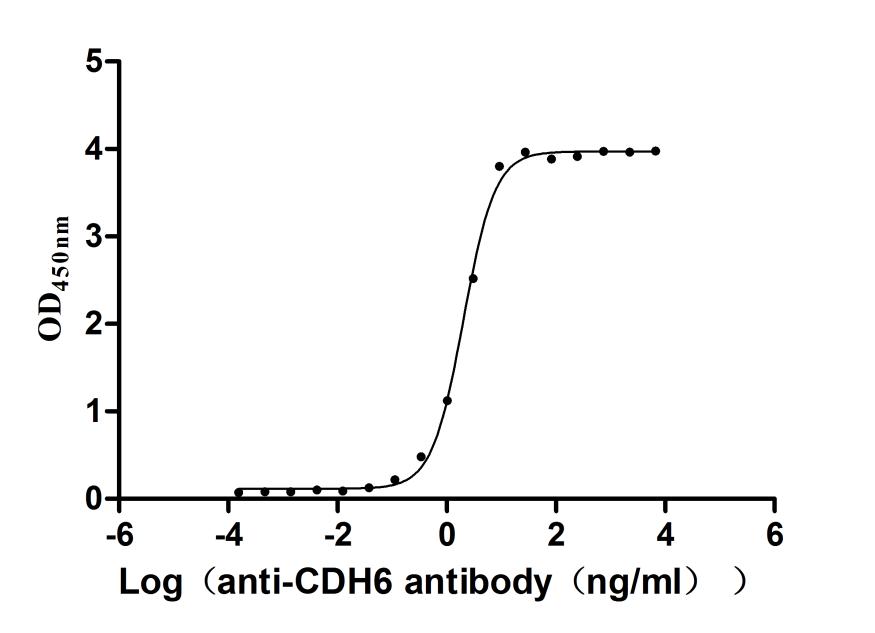
Recombinant Mouse Cadherin-6(Cdh6),partial (Active)
Express system: Mammalian cell
Species: Mus musculus (Mouse)










